Osteopetrosis is a rare hereditary bone disorder that presents in one of three forms: osteopetrosis tarda, osteopetrosis congenita and “marble bone” disease. Osteopetrosis tarda, the benign form, presents in adulthood, while the two more malignant variants, osteopetrosis congenita and marble bone disease, present in infancy and childhood, respectively. In all three forms, the main features are pathologic alteration of osteoclastic bone resorption and thickening of cortical and lamellar bones. Osteopetrosis tarda is usually discovered accidentally on routine radiographs and is often asymptomatic; however, patients may present because of related degenerative joint disease. Osteopetrosis congenita results in bone marrow failure and is almost always fatal. Marble bone disease causes short stature, cerebral calcification and mental retardation. Bone marrow transplant is the only chance for survival in patients with osteopetrosis congenita.
Osteopetrosis is a rare bone disease that may present in one of three distinct forms (Table 1). Osteopetrosis tarda, the most benign form, presents in adulthood and is often diagnosed incidentally on routine radiographs, whereas the two more malignant variants, osteopetrosis congenita and marble bone disease, present in infancy and childhood. In all three forms, the main features are pathologic alteration of osteoclastic bone resorption and thickening of cortical and lamellar bones.1
TABLE 1 Three Variants of Osteopetrosis
| Variant | Inheritance pattern | Pathophysiology | Clinical features | Age of onset | Prognosis |
|---|---|---|---|---|---|
| Osteopetrosis tarda | Autosomal dominant | Abnormal osteoclastic bone resorption | No bone marrow failure; brittle bones; increased susceptibility to fractures but with normal healing; degenerative joint disease; 50% of patients are asymptomatic | Adulthood | Good |
| Osteopetrosis congenita | Autosomal recessive | Abnormal osteoclastic bone resorption | Severe bone marrow failure; pancytopenia; bleeding; infection; failure to thrive; growth retardation; proptosis; blindness; deafness; hydrocephalus | Infancy | Poor |
| Marble bone disease | Autosomal recessive | Abnormal osteoclastic bone resorption | No bone marrow failure; renal tubular acidosis; intracranial calcifications; sensorineural hearing loss; psychomotor retardation | Childhood | Poor |
Illustrative Case
A 46-year-old man presented to the office with a mild but noticeable limp and knee pain on the right side. Examination of the knee was unremarkable, but moderate pain and restriction were elicited during active and passive range of motion of the hip. Radiographs of the hip showed generalized sclerosis and increased radiodensity of the hip bones, with irregular subcortical and articular bone resorption suggestive of osteopetrosis with concomitant degenerative joint disease. The patient subsequently underwent a total hip replacement. The patient's father and 13-year-old son were also diagnosed with osteopetrosis.
Pathophysiology
The primary underlying mechanism involved in all forms of osteopetrosis is the failure of normal osteoclastic bone resorption. This results in dense, deformed sclerotic bones1 that show typical and diagnostic patterns on radiograph (Figure 1).
FIGURE 1.
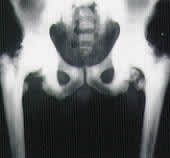
Osteopetrosis in a 46-year-old man.
Osteopetrosis tarda, the benign adult form, is inherited as an autosomal dominant trait. Patients typically are asymptomatic and have good long-term survival rates because bone marrow failure rarely occurs.1 A more common and malignant form, osteopetrosis congenita, presents in infancy and results in bone marrow failure caused by complete replacement of the marrow spaces with osteoclasts.1
An extremely rare form (only a few cases have been reported in the literature) occurs in childhood and is inherited as an autosomal recessive gene.2 The syndrome results from the absence of carbonic anhydrase isoenzyme II, an enzyme necessary for normal osteoclastic bone resorption. Major characteristics of this disorder include cerebral calcification, osteopetrosis and distal renal tubular acidosis. This form has been called marble bone disease.3
Clinical Presentation
Osteopetrosis Tarda
Osteopetrosis tarda (benign osteopetrosis) is usually detected by a family history of bone disease or as an incidental radiologic finding, and is asymptomatic in about 50 percent of cases.4 About 40 percent of patients present with fractures related to brittle osteopetrotic bones or with osteomyelitis, especially of the mandible.4 There is sufficient retention of marrow cavity for normal hematopoiesis to occur in patients with osteopetrosis tarda. In some cases, there is an elevated acid phosphatase level.4 Although patients with osteopetrosis tarda have an increased susceptibility to fractures, healing appears to proceed normally.1
Osteopetrosis Congenita
Osteopetrosis congenita (malignant osteopetrosis) presents in infancy and is associated with failure to thrive and growth retardation.4 This form of osteopetrosis is very severe and usually results in death by age two years.1 Proptosis, blindness, deafness and hydrocephalus occur in these patients as bone encroaches on the cranial foramina.5 A critical feature of osteopetrosis congenita is severe bone marrow failure, resulting in pancytopenia. Extramedullary hematopoiesis, resulting in hepatosplenomegaly and hypersplenism, may occur but cannot compensate for bone marrow failure. Thrombocytopenia, leukoerythroblastic anemia and elevated serum acid and alkaline phosphatase levels are also usually present.4 Hypocalcemia may or may not be present. Death from osteopetrosis congenita occurs as a result of severe anemia, bleeding and/or infection.4
In rare instances patients survive into adulthood. They present with severe anemia, recurrent fractures, growth retardation, deafness, blindness and massive hepatosplenomegaly.1
Marble Bone Disease
Marble bone disease, the other infantile form of osteopetrosis, is not characterized by bone marrow failure. Although survival rates are better for patients with marble bone disease than for patients with osteopetrosis congenita, the consequences of renal tubular acidosis may shorten life expectancy. Patients with marble bone disease are usually of short stature and present with intracranial calcifications, sensorineural hearing loss and psychomotor retardation.6
Clinical Management
Osteopetrosis tarda requires no treatment, except in patients who present with surgical or medical complications. Surgical treatment is sometimes essential in order to obtain the best aesthetic and functional results, such as in cases of significant alterations of facial profile7 and recurrent fractures with subsequent deformity. Surgical intervention is also necessary in some cases of severe related degenerative joint disease. Both internal8 and external9 fixations have been used with excellent results.
Several treatment modalities have been under investigation for the infantile malignant variant of osteopetrosis. Results of a recent retrospective study10 that reviewed outcomes of 69 patients who received bone marrow grafts between 1976 and 1994 indicated that bone marrow transplant is the only curative treatment for this autosomal recessive variant. This conclusion was further supported by the outcome of a study11 of eight patients who underwent allogenic bone marrow transplant from 1987 to 1992. Investigators concluded that bone marrow transplant offered a cure of both the bone marrow failure and the metabolic disturbances. Oral and external nutritional support has also been used to improve growth and enhance patient response to other treatment modalities.
Recently, several children with severe osteopetrosis were treated with 1,25-dihydroxy vitamin D in an effort to provoke nonresorbing osteoclasts to resorb bone or to cause a differentiation of mononuclear cell precursors into mature normal osteoclasts.3 Although no clinical improvements were noted, bone biopsy specimens showed evidence of increased osteoclastic bone resorption.1 This therapy is still considered experimental.
Erythropoietin was found to correct anemia and thrombocytopenia in a patient with osteopetrosis congenita who presented with “myelophthisis type” anemia that was resistant to nandrolone and low-dose corticosteroid therapy.12 Recently, a six-month trial13 of recombinant human interferon gamma in 14 patients provided evidence of long-term therapeutic benefits. These patients showed a significant increase in bone resorption, hematopoiesis (medullary) and leukocytic function. A recently published article6 reported on the follow-up of seven patients with marble bone disease who had received alkaline therapy. This treatment resulted in improvement of growth retardation but had no effect on hearing loss.
