
Addison Disease: Early Detection and Treatment Principles
Am Fam Physician. 2014;89(7):563-568
Patient information: A handout on this topic is available at https://familydoctor.org/familydoctor/en/diseases-conditions/addisons-disease.html.
Author disclosure: No relevant financial affiliations.
Primary adrenal insufficiency, or Addison disease, has many causes, the most common of which is autoimmune adrenalitis. Autoimmune adrenalitis results from destruction of the adrenal cortex, which leads to deficiencies in glucocorticoids, mineralocorticoids, and adrenal androgens. In the United States and Western Europe, the estimated prevalence of Addison disease is one in 20,000 persons; therefore, a high clinical suspicion is needed to avoid misdiagnosing a life-threatening adrenal crisis (i.e., shock, hypotension, and volume depletion). The clinical manifestations before an adrenal crisis are subtle and can include hyperpigmentation, fatigue, anorexia, orthostasis, nausea, muscle and joint pain, and salt craving. Cortisol levels decrease and adrenocorticotropic hormone levels increase. When clinically suspected, patients should undergo a cosyntropin stimulation test to confirm the diagnosis. Treatment of primary adrenal insufficiency requires replacement of mineralocorticoids and glucocorticoids. During times of stress (e.g., illness, invasive surgical procedures), stress-dose glucocorticoids are required because destruction of the adrenal glands prevents an adequate physiologic response. Management of primary adrenal insufficiency or autoimmune adrenalitis requires vigilance for concomitant autoimmune diseases; up to 50% of patients develop another autoimmune disorder during their lifetime.
More than 150 years ago, Thomas Addison described a group of patients with anemia and diseased adrenal glands at autopsy, a condition now known as primary adrenal insufficiency. Autoimmune adrenalitis is the most common cause of primary adrenal insufficiency, or Addison disease, in the United States. Less common causes include infection, hemorrhage, metastatic cancer, medication use, and adrenoleukodystrophy.
Autoimmune adrenalitis is a disorder in which the adrenal cortex is destroyed, resulting in the loss of mineralocorticoid, glucocorticoid, and adrenal androgen hormone production. Addison disease can be part of the autoimmune polyglandular syndromes (type 1 and 2), or it may present as an isolated disorder.1 This article focuses on the diagnosis and treatment of Addison disease as an isolated disorder, with a focus on the pathophysiology and treatment considerations of autoimmune adrenalitis.
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Addison disease, or primary adrenal insufficiency, is diagnosed after confirming an elevated ACTH level and an inability to stimulate cortisol levels with a cosyntropin stimulation test. | C | 12, 22 |
| Addison disease should be treated with a glucocorticoid (i.e., daily prednisone, twice daily hydrocortisone, or daily dexamethasone in situations when oral therapy is not tolerated). Treatment should be titrated to the lowest dose that relieves symptoms. [corrected] | C | 16–20 |
| Addison disease should be treated with a mineralocorticoid (i.e., daily fludrocortisone). Treatment should be titrated to keep the plasma renin activity in the upper normal range. | C | 21, 22 |
| Dehydroepiandrosterone (DHEA) therapy may improve depression symptoms and health-related quality of life in women. | B | 23 |
| Physicians should remain vigilant for the development of concomitant autoimmune disorders in patients with Addison disease. | C | 8, 28–34 |
Pathogenesis
Autoimmune adrenalitis can be divided into stages of progression2,3 (Table 13 ). As the disease develops, individuals lose adrenocortical function over a period of years. In the first three stages, the human leukocyte antigen genes confer genetic risk; an unknown precipitating event initiates antiadrenal autoimmunity; and 21-hydroxylase antibodies are produced, which predict future disease. The production of these antibodies can precede symptom onset by years to decades, and they are present in more than 90% of recent-onset cases.2,4–7 In the fourth stage, overt adrenal insufficiency develops. One of the first metabolic abnormalities to occur is an increase in plasma renin level, followed by the sequential development of other abnormalities, including a decreased response to adrenocorticotropic hormone (ACTH) stimulation in the fifth stage. If symptoms of adrenal insufficiency are present but go undiagnosed, an addisonian crisis can occur.

| Stage | Symptoms | Comments |
|---|---|---|
| 1. Genetic risk | None | HLA-B8, -DR3, and -DR4 genes confer risk |
| 2. Precipitating event starts antiadrenal autoimmunity | None | Possible environmental trigger |
| 3. 21-hydroxylase antibodies present | None | Antibodies appear before disease onset in 90% of cases |
| 4. Metabolic decompensation | Fatigue, anorexia, nausea, hyperpigmentation | Increased ACTH and decreased 8 a.m. cortisol levels; high clinical suspicion needed for diagnosis |
| 5. Decreased response to ACTH stimulation | Hypotension and shock (addisonian crisis) | Severe symptoms can be life-threatening |
Clinical Diagnosis
Because the estimated prevalence of Addison disease is one in 20,000 persons in the United States and Western Europe, a high clinical suspicion is needed to avoid misdiagnosing a life-threatening adrenal crisis.8 Signs and symptoms can be subtle and nonspecific. Patients may experience fatigue, weakness, weight loss, and gastrointestinal upset9 (Table 210 ). Symptoms are gradual and worsen over a period of years, making early diagnosis difficult.10 The symptoms relate to the degree of cortisol, mineralocorticoid, and adrenal androgen deficiency at the time of presentation. Addison disease is usually diagnosed after a significant stress or illness unmasks cortisol and mineralocorticoid deficiency, presenting as shock, hypotension, and volume depletion (adrenal or addisonian crisis).11 Cortisol and aldosterone deficiencies contribute to hypotension, orthostasis, and shock; however, adrenal crisis is more likely to occur in primary adrenal insufficiency compared with secondary adrenal insufficiency.

| Sign or symptom | Prevalence (%) |
|---|---|
| Anorexia | 100 |
| Weakness, fatigue | 100 |
| Hyperpigmentation | 94 |
| Gastrointestinal symptoms (e.g., nausea, vomiting, abdominal pain, constipation, diarrhea) | 92 |
| Hypotension (systolic blood pressure < 110 mm Hg) | ~90 |
| Salt cravings | 16 |
| Postural dizziness | 12 |
| Vitiligo | 10 to 20 |
| Muscle or joint pain | ~10 |
Hyperpigmentation is the physical finding most characteristic of Addison disease, arising from continual stimulation of the corticotrophs in the anterior pituitary. Specifically, it results from cross-reactivity between the ACTH produced by the corticotrophs and the melanocortin 1 receptor on keratinocytes. Hyperpigmentation is usually generalized over the entire body and can be found in palmar creases, buccal mucosa, vermilion border of the lips, and around scars and nipples. It is not a feature of secondary adrenal insufficiency because of the lack of increased ACTH in these patients.
Diagnosis
METABOLIC TESTS
The goal of laboratory testing is to document a low cortisol level and determine whether the adrenal insufficiency is primary or secondary, as outlined in Figure 1. Low serum cortisol levels at 8 a.m. (less than 3 mcg per dL [83 nmol per L]) suggest adrenal insufficiency, as do levels.12 Hyponatremia can be attributed to cortisol and mineralocorticoid deficiencies, whereas hyperkalemia is attributed solely to a lack of mineralocorticoids.
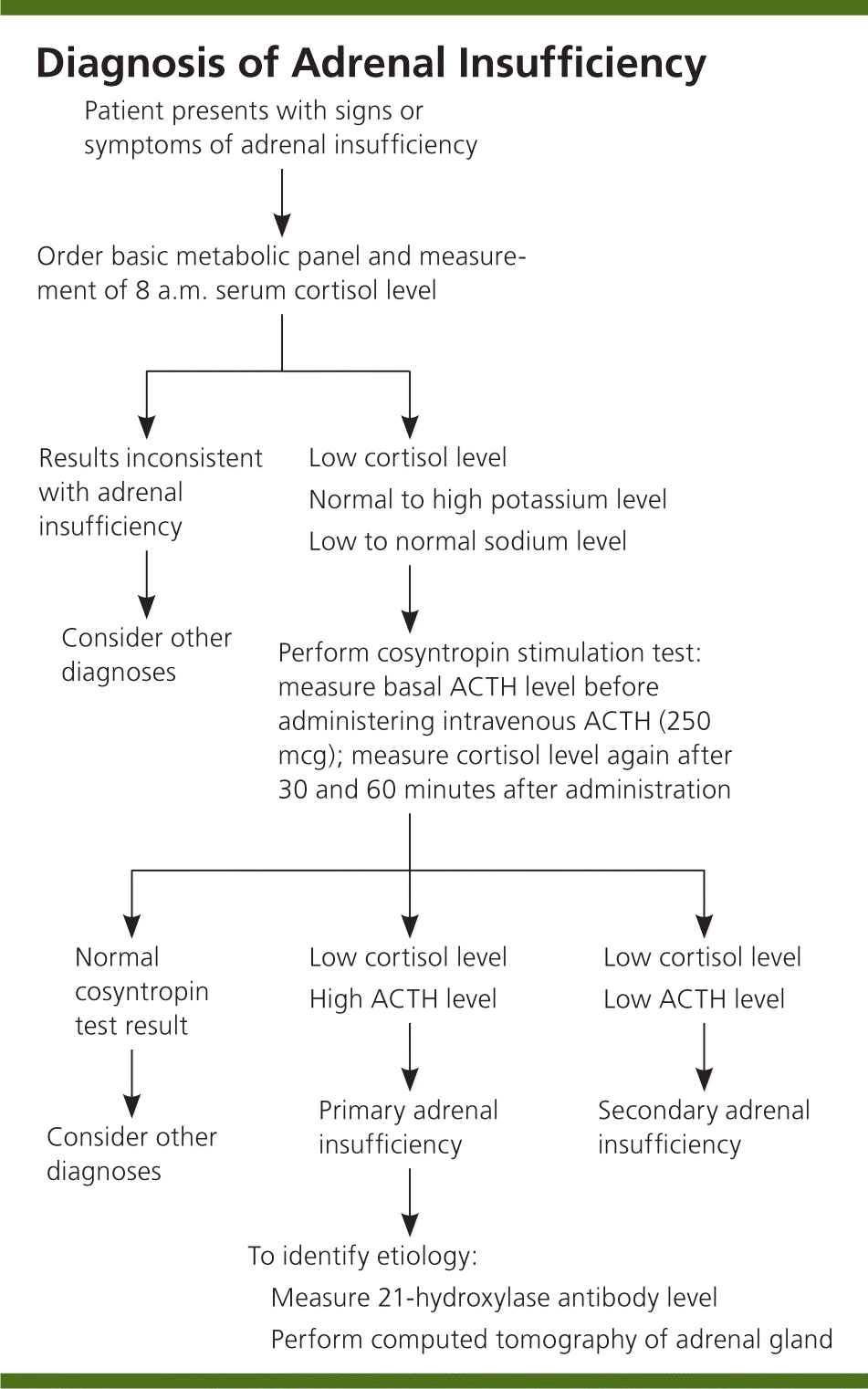
Because the adrenal hormones are gradually lost over years to decades, the levels vary. One of the first indications that there is adrenal cortex dysfunction is an elevated plasma renin level.13 A rise in ACTH levels is concomitant with the loss of adrenal hormones. Yearly monitoring of ACTH levels in at-risk individuals shows that measurements greater than 50 pg per mL (11 pmol per L), which exceed the upper limit of normal, are indicative of cortisol deficiency.7 A cosyntropin stimulation test is the first-line test for diagnosing adrenal insufficiency. The serum cortisol, plasma ACTH, plasma aldosterone, and plasma renin levels should be measured before administering 250 mcg of ACTH. At 30 and 60 minutes after intravenous ACTH administration, the serum cortisol level should be measured again. A normal response occurs with peak cortisol levels greater than 18 to 20 mcg per dL (497 to 552 nmol per L); a smaller or absent response is diagnostic for adrenal insufficiency.14,15
IMMUNOLOGIC TESTS
Measurement of 21-hydroxylase antibody levels helps discern the cause of Addison disease. The 21-hydroxylase enzyme is necessary for cortisol synthesis in the adrenal cortex; antibodies directed against the enzyme are specific for autoimmune adrenalitis and are detectable before symptom onset.
IMAGING
Radiographic imaging is also helpful in determining the cause of Addison disease, but it is relatively nonspecific in patients with autoimmune destruction. It is important to make a biochemical diagnosis of adrenal insufficiency before radiographic imaging. Computed tomography demonstrates small adrenal glands in patients with autoimmune adrenal destruction. In other causes of Addison disease, computed tomography may show hemorrhage, calcification associated with tuberculosis infection, or masses in the adrenal gland. However, computed tomography is not necessary to diagnose adrenal insufficiency.
Treatment
HORMONE THERAPY
Treatment for Addison disease consists of lifelong hormone therapy with glucocorticoids and mineralocorticoids16 (Table 3). To date, there is no therapy available to stop the underlying immune destruction of the adrenal cortex. Generally, glucocorticoid replacement includes oral prednisone or hydrocortisone.17 Prednisone can be taken once daily, whereas hydrocortisone is divided into two or three doses per day.18–20 Mineralocorticoids are replaced with fludrocortisone at a dose sufficient to keep the plasma renin level in the upper limit of the normal range.21,22
Men who have Addison disease do not need replacement with androgens because their testes are able to produce adequate testosterone levels; however, women can benefit from androgen replacement because the adrenals are the main source of androgen production in women. A meta-analysis of 10 randomized placebo-controlled trials found that dehydroepiandrosterone (DHEA) supplementation resulted in small improvements in health-related quality of life and depression in women with adrenal insufficiency.23

| Medication | Dosage | Comments | Monitoring |
|---|---|---|---|
| Glucocorticoids | |||
| Prednisone | 3 to 5 mg once daily | Use stress doses for illness, surgical procedures, and hospitalization | Symptoms of adrenal insufficiency; low to normal plasma adrenocorticotropic hormone levels indicate over-replacement |
| Hydrocortisone | 15 to 25 mg divided into two or three doses per day | Use stress doses for illness, surgical procedures, and hospitalization | |
| Dexamethasone | 0.5 mg once daily | Use intramuscular dose for emergencies and when unable to tolerate oral intake | |
| Mineralocorticoid | |||
| Fludrocortisone | 0.05 to 0.2 mg once daily | Dosage may need to increase to 0.2 mg per day in the summer because of salt loss from perspiration | Blood pressure; serum sodium and potassium levels; plasma renin activity in the upper normal range |
| Androgen | |||
| Dehydroepiandrosterone (DHEA) | 25 to 50 mg once daily | Available as an over-the-counter supplement; can improve mood and quality of life in women | Libido, mood, and sense of well-being |
STRESS DOSING OF GLUCOCORTICOIDS
Patients should be counseled about the need for stress-dose glucocorticoids for illnesses and before surgical procedures because destruction of the adrenal glands prevents an adequate physiologic response to stress.24 There are many expert recommendations for stress dosing of steroids based on the degree of stress; clinical trials comparing different approaches are lacking in the literature. In our practice, we use a stress-dose strategy for outpatient procedures (e.g., colonoscopy, upper endoscopy) and invasive dental procedures (e.g., root canal) that patients can implement easily. This involves a dose of glucocorticoids three times the maintenance dose the day of the procedure and two days after (i.e., three times three rule for stress-dose glucocorticoids).
For minor illnesses such as influenza or viral gastroenteritis, the patient can take three times the steroid dose during the illness and resume normal dosing after resolution of symptoms. Patients should also have an injectable form of glucocorticoid (intramuscular dexamethasone) available in cases of nausea, vomiting, or other situations when oral intake is not possible. Mineralocorticoid replacement generally does not need to be changed for illness or procedures. However, the dose may need to be adjusted in the summer months when there is salt loss from excessive perspiration.
TREATMENT CAVEATS
Thyroid hormone therapy in persons with undiagnosed Addison disease may precipitate an adrenal crisis because the thyroid hormone increases the hepatic clearance of cortisol. In addition, patients with a new diagnosis can have a reversible increase in thyroid-stimulating hormone levels because glucocorticoids inhibit secretion.25,26 Glucocorticoid replacement can result in the normalization of thyroid-stimulating hormone levels less than 30 mIU per L. In individuals with type 1 diabetes mellitus, unexplained hypoglycemia and decreasing insulin requirements may be the initial signs of Addison disease.27
TREATMENT OF CONFIRMED ADDISON DISEASE
Patients with Addison disease should be treated in conjunction with an endocrinologist and be monitored on a regular basis for appropriate hormone therapy (Table 3). Glucocorticoid doses should be titrated to the lowest tolerated dose that controls symptoms to minimize the adverse effects of excess glucocorticoid. It is important to instruct patients to learn the proper guidelines for stress dosing of glucocorticoids, to have an injectable form of glucocorticoid available, and to wear an adrenal insufficiency medical alert identification.
Approximately 50% of persons with Addison disease caused by autoimmune adrenalitis develop another autoimmune disorder during their lifetime, necessitating lifelong vigilance for associated autoimmune conditions.28,29 Table 4 outlines concomitant autoimmune disorders and their relative prevalence, as well as appropriate autoantibodies and metabolic tests for patients with Addison disease who develop signs and symptoms of one of these disorders.8,28–34 Of note, 10% of women with Addison disease experience autoimmune premature ovarian failure, or primary ovarian insufficiency, in their reproductive years with signs and symptoms of estrogen deficiency (e.g., amenorrhea, flushing, fatigue, poor concentration).34 It is appropriate to offer these patients evaluation and counseling on other options for building a family.35

| Disease | Lifetime prevalence (%) | Appropriate diagnostic tests |
|---|---|---|
| Autoimmune thyroid disease (Hashimoto disease or Graves disease)8,28–32 | 22 | Thyroid-stimulating hormone, thyroid peroxidase antibody, and thyroid-stimulating immunoglobulin levels |
| Celiac disease33 | 12 | Tissue transglutaminase antibody level |
| Type 1 diabetes mellitus8,28–30,32 | 11 | A1C, fasting blood glucose, and islet autoantibody levels |
| Hypoparathyroidism8,28–30,32 | 10 | Calcium and parathyroid hormone levels |
| Primary ovarian insufficiency34 | 10 | Follicle-stimulating hormone level |
| Pernicious anemia8,28,29 | 5 | Complete blood count, vitamin B12 level, and parietal cell antibody level |
| Primary gonadal failure (testes) 29 | 2 | Testosterone, follicle-stimulating hormone, and luteinizing hormone levels |
| None8,28–34 | 50 | — |
Online resources for physicians and patients can be found at the National Adrenal Diseases Foundation (http://www.nadf.us), the Addison's Disease Self Help Group (http://www.addisons.org.uk), and the Canadian Addison Society (addisonsociety.ca). [corrected]
Data Sources: Articles were searched using the PubMed and Cochrane databases, and obtained from the Essential Evidence Plus summary provided by the journal editors. The search terms included Addison disease, autoimmune primary adrenal insufficiency, cosyntropin stimulation testing, glucocorticoid treatment, mineralocorticoid treatment, DHEA treatment, and the immunology of Addison disease. Articles with abstracts that were published in English within the past five years were the primary focus; older literature on the signs, symptoms, and diagnosis of Addison disease were cited as appropriate. Search dates: December 2011 and October 2013.
