
Am Fam Physician. 2021;103(8):481-492
Patient information: See related handout on hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorder, written by the authors of this article.
Author disclosure: No relevant financial affiliations.
Hypermobile Ehlers-Danlos syndrome (EDS) and hypermobility spectrum disorders are the most common symptomatic joint hypermobility conditions seen in clinical practice. The 2017 International Classification of the Ehlers-Danlos syndromes replaced previous terms for symptomatic joint hypermobility with hypermobile EDS and introduced the term hypermobility spectrum disorders for patients not meeting diagnostic criteria for hypermobile EDS. Both are diagnosed by applying the 2017 diagnostic criteria, which also excludes other less common conditions presenting with joint hypermobility such as other forms of EDS and heritable connective tissue disorders. Hypermobile EDS is inherited in an autosomal dominant pattern, but it does not have a known genetic mutation to help with diagnosis. Clinical features of hypermobile EDS include joint hypermobility, skin findings, and joint pains or recurrent dislocations. Hypermobile EDS and, less commonly, hypermobility spectrum disorders may also be associated with several extra-articular symptoms, including anxiety disorders, chronic pain, fatigue, orthostatic intolerance, functional gastrointestinal disorders, and pelvic and bladder dysfunction. The central goals of therapy are managing symptoms, preventing joint injury, and educating patients about their condition. Based on limited evidence, patients with hypermobile EDS/hypermobility spectrum disorders may benefit from physical and occupational therapy, psychological support, and self-management. Primary care physicians play a key role not only in initial recognition, diagnosis, and patient education, but by virtue of their ongoing relationship they can also help oversee and coordinate the multidisciplinary team many of these patients require.

| Clinical recommendation | Evidence rating | Comments |
|---|---|---|
| Suspect hypermobile EDS/hypermobility spectrum disorders in patients with joint hypermobility and associated symptoms such as joint pain or dislocations and typical skin findings, arthralgias, recurrent hernias, marfanoid habitus, or family history of EDS.1 | C | Expert opinion from 2017 International Classification of the Ehlers-Danlos syndromes |
| Assess joint hypermobility in patients suspected of having hypermobile EDS/hypermobility spectrum disorders with a Beighton score and a validated five-part questionnaire.1,41,43 | C | Disease-oriented outcomes and expert opinion |
| Early multidisciplinary treatment that includes physical, occupational, and cognitive behavior therapy; orthotics; and community and specialty support may optimize outcomes in patients with joint hypermobility symptoms.46–48,58,59 | C | Expert opinion and several case series studies |
Definitions
“Ehlers-Danlos syndromes (EDS) … are a group of inherited connective tissue disorders caused by abnormalities in the structure, production, and/or processing of collagen. The new classification, from 2017, includes 13 subtypes of EDS.”3 Table 1 describes these subtypes.1,4,5 Joint hypermobility is a feature common among many EDS subtypes and other heritable connective tissue disorders. Joint hypermobility is defined as the ability of a joint to move “beyond normal limits along physiological axes.”4 Joint hypermobility can involve a few or many joints and may be entirely asymptomatic. Generalized joint hypermobility is more likely to be associated with a genetic syndrome than localized joint hypermobility. The exception is hypermobile EDS, which is the most common EDS variant, representing 80% to 90% of EDS cases,2 with clinical features including joint hypermobility, skin findings (Figure 1), and joint pains or recurrent dislocations (Figure 26 and Figure 31,7). The 2017 classification introduced stricter criteria for hypermobile EDS than previously available to distinguish patients with hypermobile EDS from those most likely to have a diagnosable genetic syndrome.1 For patients with symptomatic joint hypermobility satisfying neither the new hypermobile EDS criteria nor another specific condition, the 2017 classification introduced hypermobility spectrum disorders.1
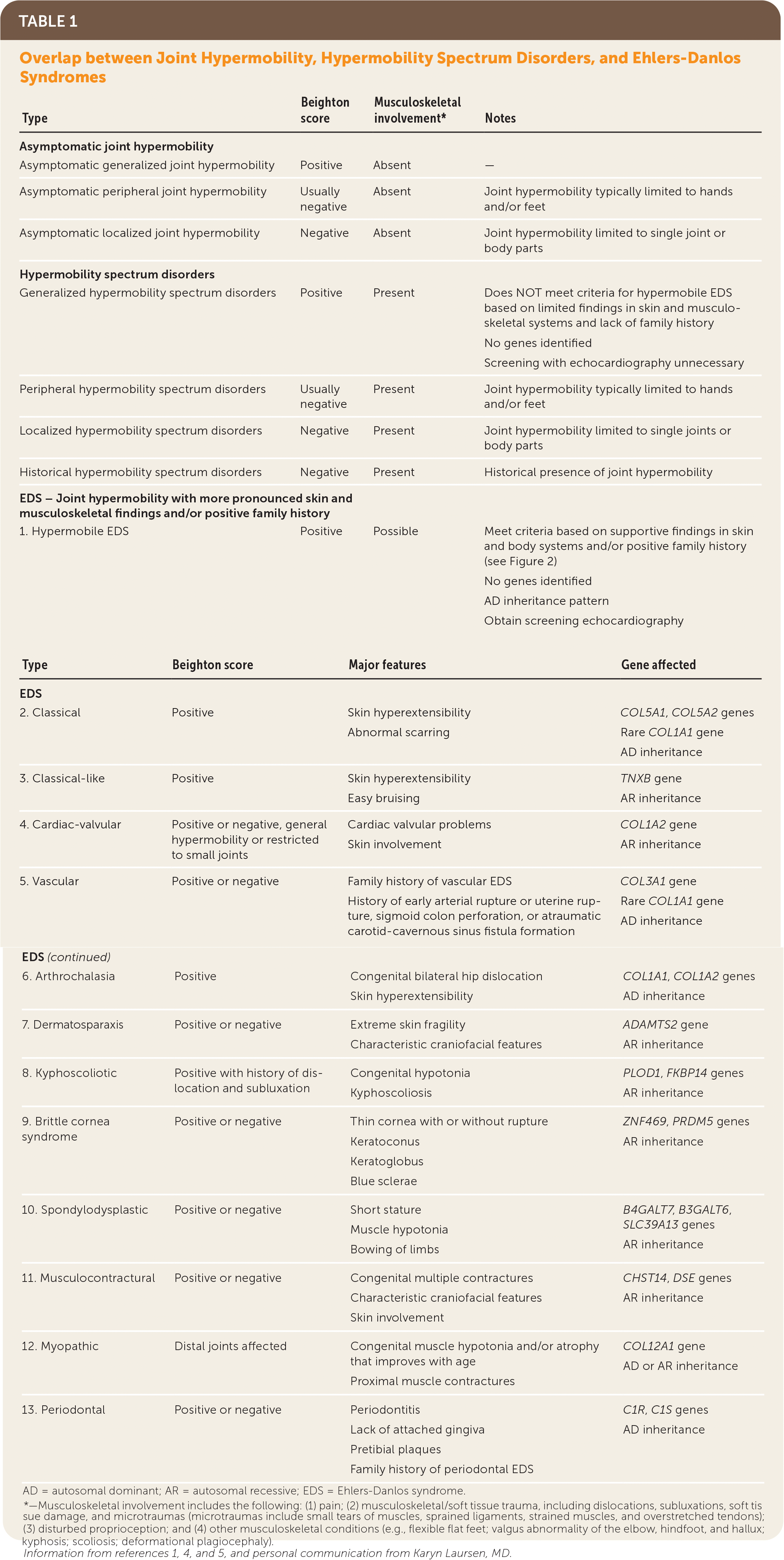
| Type | Beighton score | Musculoskeletal involvement* | Notes |
|---|---|---|---|
| Asymptomatic joint hypermobility | |||
| Asymptomatic generalized joint hypermobility | Positive | Absent | — |
| Asymptomatic peripheral joint hypermobility | Usually negative | Absent | Joint hypermobility typically limited to hands and/or feet |
| Asymptomatic localized joint hypermobility | Negative | Absent | Joint hypermobility limited to single joint or body parts |
| Hypermobility spectrum disorders | |||
| Generalized hypermobility spectrum disorders | Positive | Present | Does NOT meet criteria for hypermobile EDS based on limited findings in skin and musculoskeletal systems and lack of family history No genes identified Screening with echocardiography unnecessary |
| Peripheral hypermobility spectrum disorders | Usually negative | Present | Joint hypermobility typically limited to hands and/or feet |
| Localized hypermobility spectrum disorders | Negative | Present | Joint hypermobility limited to single joints or body parts |
| Historical hypermobility spectrum disorders | Negative | Present | Historical presence of joint hypermobility |
| EDS – Joint hypermobility with more pronounced skin and musculoskeletal findings and/or positive family history | |||
| 1. Hypermobile EDS | Positive | Possible | Meet criteria based on supportive findings in skin and body systems and/or positive family history (see Figure 2) No genes identified AD inheritance pattern Obtain screening echocardiography |
| Type | Beighton score | Major features | Gene affected |
| EDS | |||
| 2. Classical | Positive | Skin hyperextensibility Abnormal scarring | COL5A1, COL5A2 genes Rare COL1A1 gene AD inheritance |
| 3. Classical-like | Positive | Skin hyperextensibility Easy bruising | TNXB gene AR inheritance |
| 4. Cardiac-valvular | Positive or negative, general hypermobility or restricted to small joints | Cardiac valvular problems Skin involvement | COL1A2 gene AR inheritance |
| 5. Vascular | Positive or negative | Family history of vascular EDS History of early arterial rupture or uterine rupture, sigmoid colon perforation, or atraumatic carotid-cavernous sinus fistula formation | COL3A1 gene Rare COL1A1 gene AD inheritance |
| 6. Arthrochalasia | Positive | Congenital bilateral hip dislocation Skin hyperextensibility | COL1A1, COL1A2 genes AD inheritance |
| 7. Dermatosparaxis | Positive or negative | Extreme skin fragility Characteristic craniofacial features | ADAMTS2 gene AR inheritance |
| 8. Kyphoscoliotic | Positive with history of dislocation and subluxation | Congenital hypotonia Kyphoscoliosis | PLOD1, FKBP14 genes AR inheritance |
| 9. Brittle cornea syndrome | Positive or negative | Thin cornea with or without rupture Keratoconus Keratoglobus Blue sclerae | ZNF469, PRDM5 genes AR inheritance |
| 10. Spondylodysplastic | Positive or negative | Short stature Muscle hypotonia Bowing of limbs | B4GALT7, B3GALT6, SLC39A13 genes AR inheritance |
| 11. Musculocontractural | Positive or negative | Congenital multiple contractures Characteristic craniofacial features Skin involvement | CHST14, DSE genes AR inheritance |
| 12. Myopathic | Distal joints affected | Congenital muscle hypotonia and/or atrophy that improves with age Proximal muscle contractures | COL12A1 gene AD or AR inheritance |
| 13. Periodontal | Positive or negative | Periodontitis Lack of attached gingiva Pretibial plaques Family history of periodontal EDS | C1R, C1S genes AD inheritance |

Patients with hypermobility spectrum disorders are distinct from those with hypermobile EDS and other syndromes with joint hypermobility in that their symptoms are primarily musculoskeletal; however, limited extra-articular involvement may be seen.4 All previous terms, including EDS type III, EDS hypermobility type, hypermobility syndrome, joint hypermobility syndrome, and benign joint hypermobility syndrome, should no longer be used.4 At one time, these earlier named diagnoses were thought to represent distinct entities, but subsequent studies finding broad overlap of these older named conditions within families demonstrated that they were the same entity.4 In a more recent study, nearly all patients with one of the earlier, now outdated, diagnoses fulfilled either hypermobile EDS or hypermobility spectrum disorders criteria.8 Because they are the most common symptomatic hypermobility conditions, their evaluation and management are the focus of this article; the terms hypermobile EDS and hypermobility spectrum disorders will be used except when clarity dictates reference to an older diagnostic term.
Epidemiology and Pathogenesis
The exact prevalence of hypermobile EDS/hypermobility spectrum disorders is unknown. The best estimates of the population prevalence of these conditions are derived from studies in national or patient registries from Sweden and Wales, United Kingdom, using diagnostic codes for EDS and joint hypermobility syndrome, the latter a prior term for hypermobile EDS, as discussed previously.9,10 The combined hypermobile EDS/hypermobility spectrum disorders prevalence would be expected to be lower than the 0.13% to 0.19% prevalence that these two studies found for all EDS and joint hypermobility syndrome codes combined.9,10 This prevalence equates to about seven to 10 patients out of a 5,000-patient panel. Another estimate of combined hypermobile EDS/hypermobility spectrum disorders prevalence from a U.K. population survey found that 3.4% of adults endorsed hypermobility and chronic widespread pain using validated instruments.11
Generalized joint hypermobility, a diagnostic criterion for hypermobile EDS, is more common than hypermobile EDS/hypermobility spectrum disorders because patients with generalized joint hypermobility may be asymptomatic. When assessed in student population samples using 2017 criteria, 4% to 11% of children three to 19 years of age had generalized joint hypermobility.12–17 The percentage of people with generalized joint hypermobility who are eventually diagnosed with hypermobile EDS/hypermobility spectrum disorders is unknown.
Hypermobile EDS is the only EDS subtype for which a genetic mutation has not been discovered. Hypermobile EDS is considered to be inherited in an autosomal dominant manner with incomplete penetrance. The pathogenesis of hypermobile EDS and hypermobility spectrum disorders is still being unraveled but involves muscle and tendon laxity,18 reduced proprioception,19 significantly disordered connective tissue structure, and alterations in gene expression.20
Clinical Presentation
Hypermobile EDS and hypermobility spectrum disorders exhibit a complex range of signs and symptoms of varying degrees and combinations that make these conditions difficult to recognize. Common presenting features of hypermobile EDS are listed in Table 2.1,2,21 The prevalence of generalized joint hypermobility declines with age,2 and this decline is considered by the 2017 hypermobile EDS criteria by incorporating historical questions for patients with subthreshold joint hypermobility.1,7 The strongest systemic findings associated with hypermobile EDS include anxiety disorders, chronic pain, fatigue, orthostatic intolerance, functional gastrointestinal disorders, and pelvic and bladder dysfunction.22–26 Table 3 describes the symptoms and physical findings commonly associated with hypermobile EDS.2,22,23,26–38
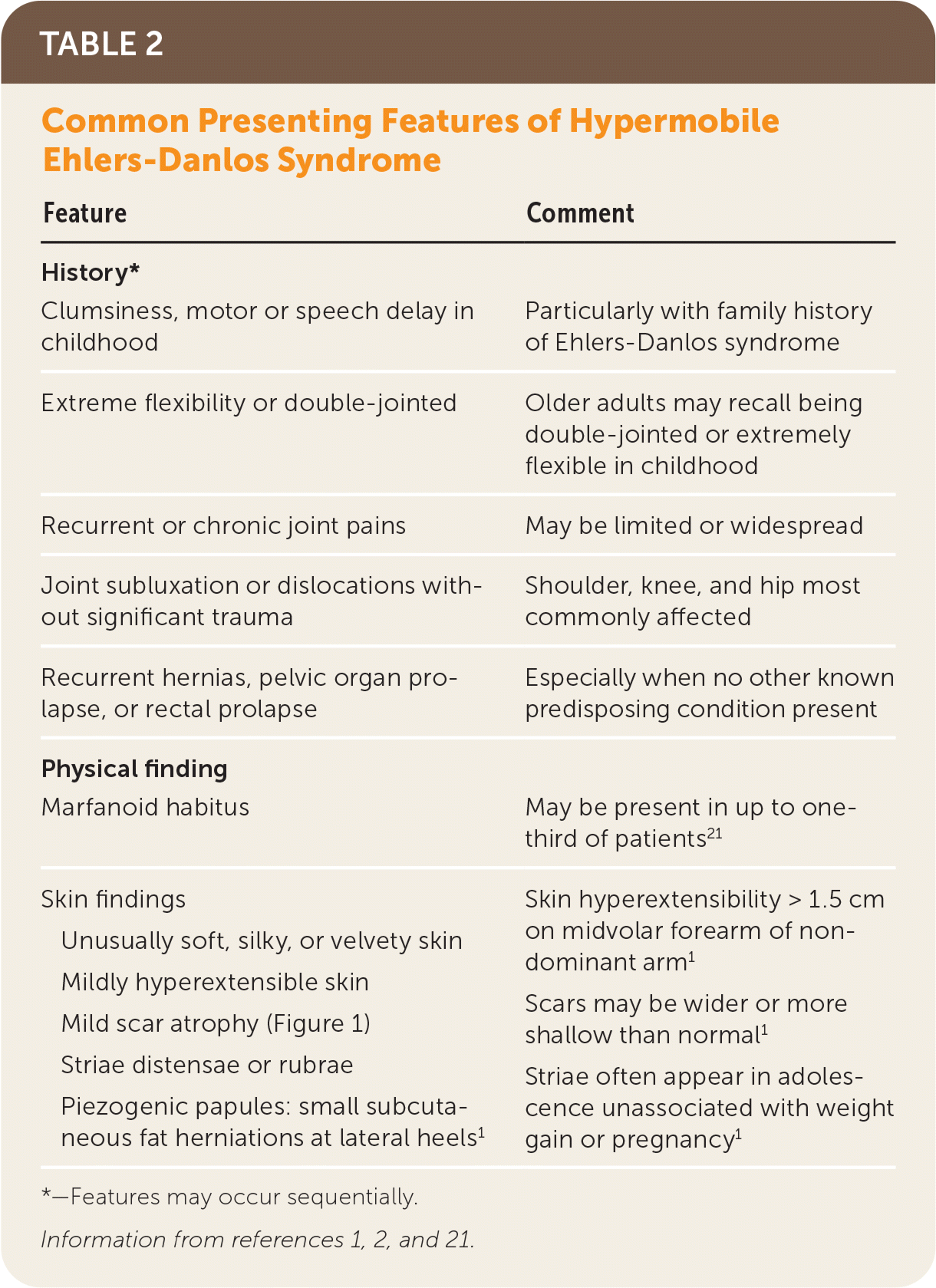
| Feature | Comment |
|---|---|
| History* | |
| Clumsiness, motor or speech delay in childhood | Particularly with family history of Ehlers-Danlos syndrome |
| Extreme flexibility or double-jointed | Older adults may recall being double-jointed or extremely flexible in childhood |
| Recurrent or chronic joint pains | May be limited or widespread |
| Joint subluxation or dislocations without significant trauma | Shoulder, knee, and hip most commonly affected |
| Recurrent hernias, pelvic organ prolapse, or rectal prolapse | Especially when no other known predisposing condition present |
| Physical finding | |
| Marfanoid habitus | May be present in up to one-third of patients21 |
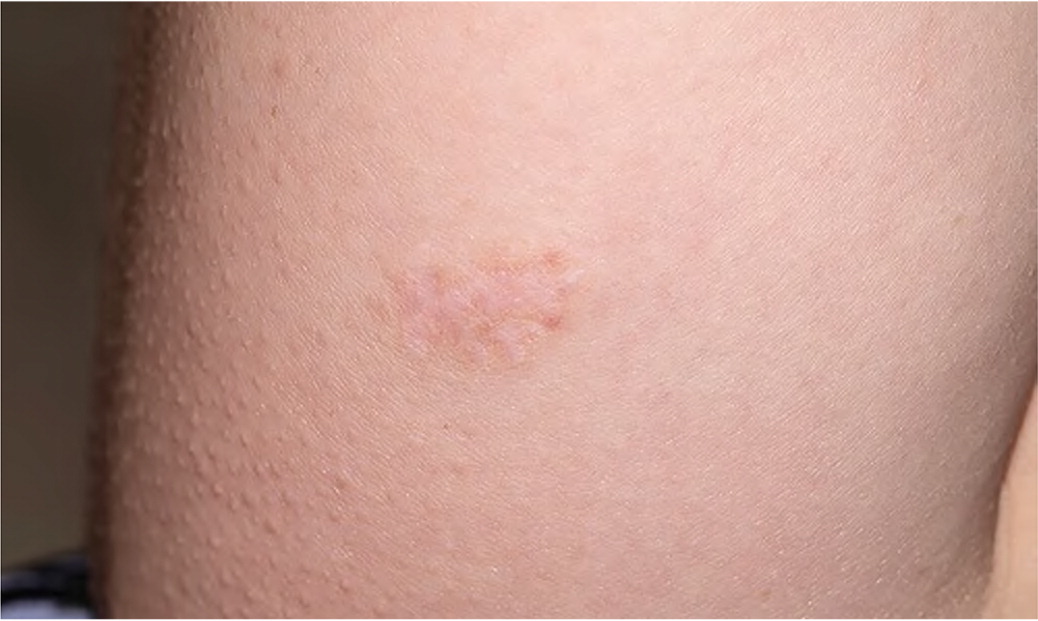
| Skin findings Unusually soft, silky, or velvety skin Mildly hyperextensible skin Mild scar atrophy (Figure 1) Striae distensae or rubrae Piezogenic papules: small subcutaneous fat herniations at lateral heels1 | Skin hyperextensibility > 1.5 cm on midvolar forearm of nondominant arm1 Scars may be wider or more shallow than normal1 Striae often appear in adolescence unassociated with weight gain or pregnancy1 |
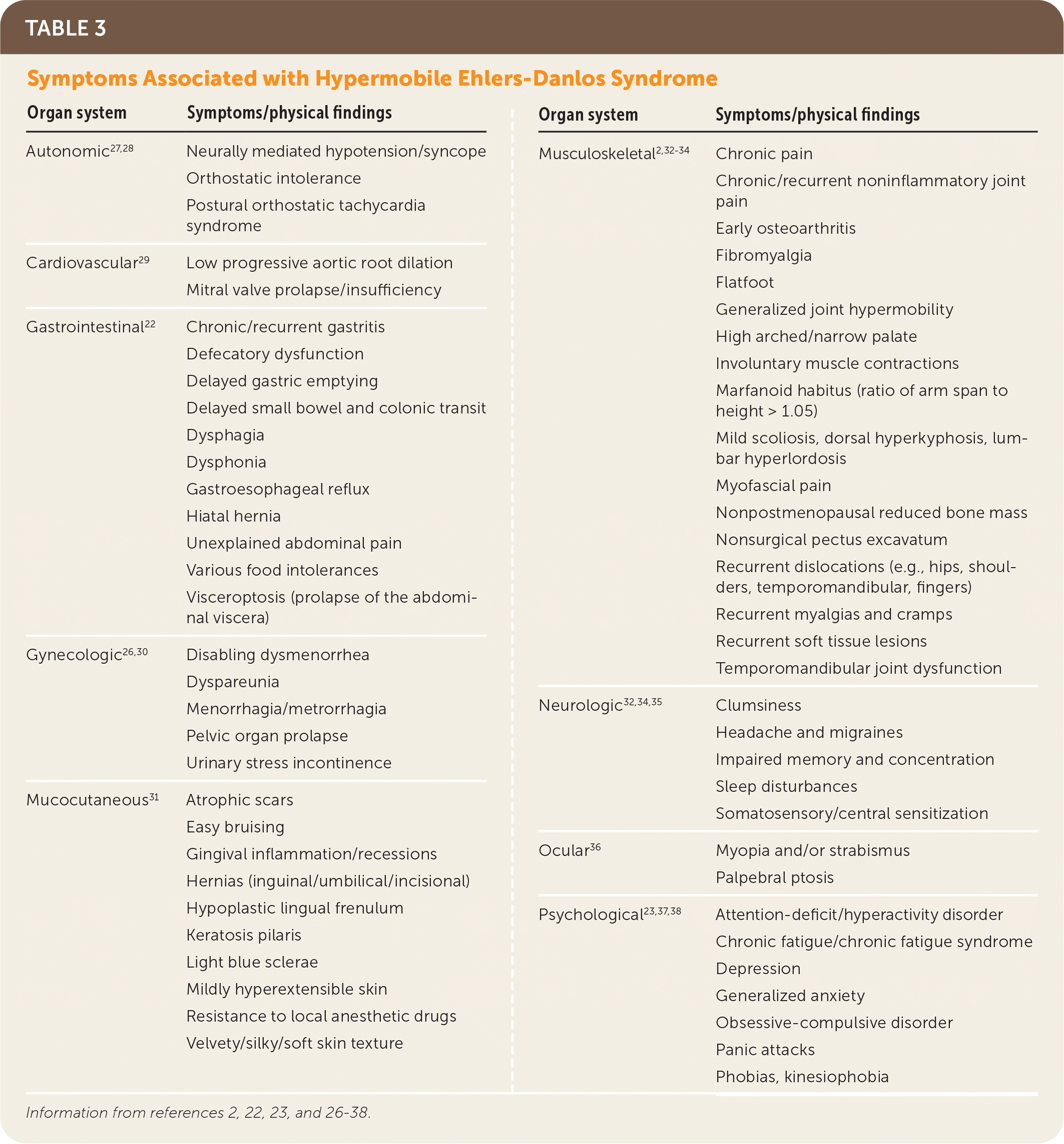
| Organ system | Symptoms/physical findings |
|---|---|
| Autonomic27,28 | Neurally mediated hypotension/syncope Orthostatic intolerance Postural orthostatic tachycardia syndrome |
| Cardiovascular29 | Low progressive aortic root dilation Mitral valve prolapse/insufficiency |
| Gastrointestinal22 | Chronic/recurrent gastritis Defecatory dysfunction Delayed gastric emptying Delayed small bowel and colonic transit Dysphagia Dysphonia Gastroesophageal reflux Hiatal hernia Unexplained abdominal pain Various food intolerances Visceroptosis (prolapse of the abdominal viscera) |
| Gynecologic26,30 | Disabling dysmenorrhea Dyspareunia Menorrhagia/metrorrhagia Pelvic organ prolapse Urinary stress incontinence |
| Mucocutaneous31 | Atrophic scars Easy bruising Gingival inflammation/recessions Hernias (inguinal/umbilical/incisional) Hypoplastic lingual frenulum Keratosis pilaris Light blue sclerae Mildly hyperextensible skin Resistance to local anesthetic drugs Velvety/silky/soft skin texture |
| Musculoskeletal2,32–34 | Chronic pain Chronic/recurrent noninflammatory joint pain Early osteoarthritis Fibromyalgia Flatfoot Generalized joint hypermobility High arched/narrow palate Involuntary muscle contractions Marfanoid habitus (ratio of arm span to height > 1.05) Mild scoliosis, dorsal hyperkyphosis, lumbar hyperlordosis Myofascial pain Nonpostmenopausal reduced bone mass Nonsurgical pectus excavatum Recurrent dislocations (e.g., hips, shoulders, temporomandibular, fingers) Recurrent myalgias and cramps Recurrent soft tissue lesions Temporomandibular joint dysfunction |
| Neurologic32,34,35 | Clumsiness Headache and migraines Impaired memory and concentration Sleep disturbances Somatosensory/central sensitization |
| Ocular36 | Myopia and/or strabismus Palpebral ptosis |
| Psychological23,37,38 | Attention-deficit/hyperactivity disorder Chronic fatigue/chronic fatigue syndrome Depression Generalized anxiety Obsessive-compulsive disorder Panic attacks Phobias, kinesiophobia |
Diagnostic Evaluation
CLINICAL FEATURES
The diagnosis of hypermobile EDS should be considered in patients with clinical features noted in Table 2.1,2,21 Patients with systemic manifestations (Table 32,22,23,26–38) and a history of joint hypermobility or arthralgias also may have hypermobile EDS or a related disorder that was overlooked or previously misdiagnosed.1 A U.K. patient survey found the median time to diagnosis was 10 years.39
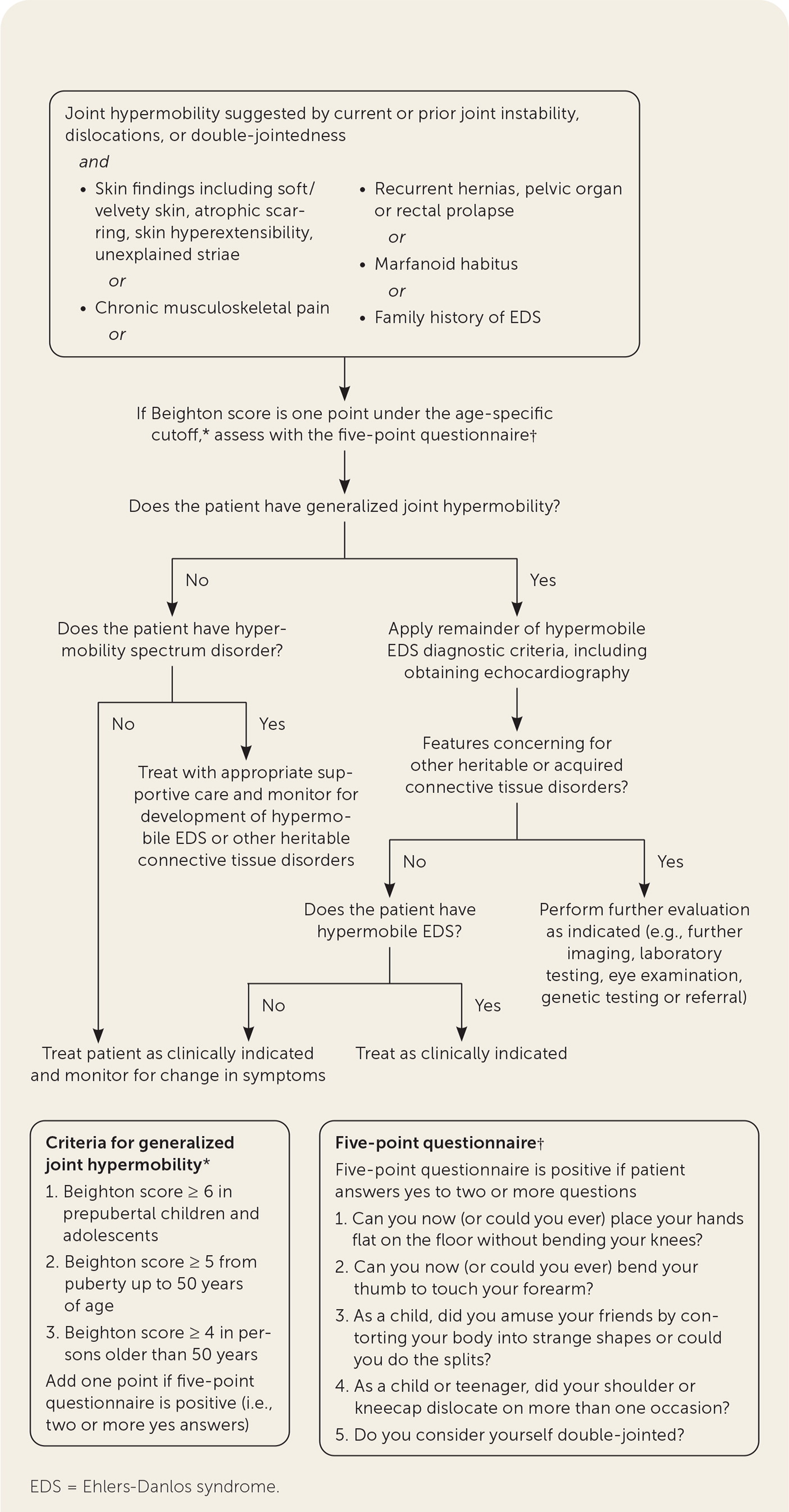
The diagnosis of hypermobile EDS/hypermobility spectrum disorders is made by medical history, physical examination, and exclusion of other conditions that present with musculoskeletal hypermobility.2,40,41 Diagnostic criteria for hypermobile EDS are listed in Figure 2.6 Table 1 outlines criteria for hypermobility spectrum disorders when patients meet neither criteria for hypermobile EDS nor another specific condition.1,4,5 A differential diagnosis for joint hypermobility appears in Table 4.1,4,21,42 Taking a careful family history that inquires about joint hypermobility, musculoskeletal symptoms, aneurysms, and genetic conditions is essential. Once hypermobile EDS is suspected, the physician should determine the degree and pattern of hypermobility using a validated tool known as the Beighton score1,40,41,43 (Table 51,44). The Beighton score incorporates five maneuvers to calculate a score between 0 and 9. The 2017 hypermobile EDS criteria in Figure 2 specify that if the Beighton score is one point below age-specific and sex-specific cutoffs for generalized joint hypermobility, the next step is to administer a validated five-part questionnaire to help determine whether the patient has generalized joint hypermobility, a necessary criterion for the diagnosis of hypermobile EDS.6,7 Patients without generalized joint hypermobility may still have hypermobility spectrum disorders. Figure 3 suggests an evaluation strategy for patients suspected of having hypermobile EDS or hypermobility spectrum disorders.1,7

| Condition | Distinguishing features |
|---|---|
| Acquired conditions including diffuse degenerative disorders of muscles, joints, or nerves; hypothyroidism; or malnutrition | Suggestive medical history |
| Chromosomal and genomic disorders including Down syndrome, aneuploidies (47, XXY; 47, XXX), and several microdeletion and microduplication syndromes | Dysmorphic features Hypogonadism |
| Hereditary cutis laxa42: multiple subtypes | Loose, inelastic skin |
| Hereditary myopathies: Bethlem, Ullrich, and others | Hypotonia and weakness Joint hyperlaxity and contractures |
| Loeys-Dietz syndrome | Arterial tortuosity and aortic aneurysms Cleft palate/bifid uvula Hypertelorism Hypotonia |
| Marfan syndrome, Beals syndrome, MASS phenotype, and arterial tortuosity syndrome | Ectopia lentis in Marfan syndrome Mitral valve prolapse in MASS phenotype Progressive ascending aortic dilation Tortuous medium and large arteries in arterial tortuosity syndrome |
| Multiple congenital anomaly or intellectual disability disorders including RASopathies, Kabuki make-up syndrome, and FG syndrome Fragile X syndrome | Multiple congenital anomalies or intellectual disabilities |
| Other variants of Ehlers-Danlos syndrome including classical, vascular, and rarer forms | Acrogeria* Arterial, viscus, or lung rupture Features specific to rarer types of Ehlers-Danlos syndrome in Table 1 Muscle weakness Pronounced skin hyperextensibility Unusual skin fragility, papyraceous or hemosideric scars |
| Skeletal dysplasias: osteogenesis imperfecta type I, Larsen syndrome, Desbuquois syndrome, and others | Blue sclerae Bone fragility Neonatal joint dislocations |
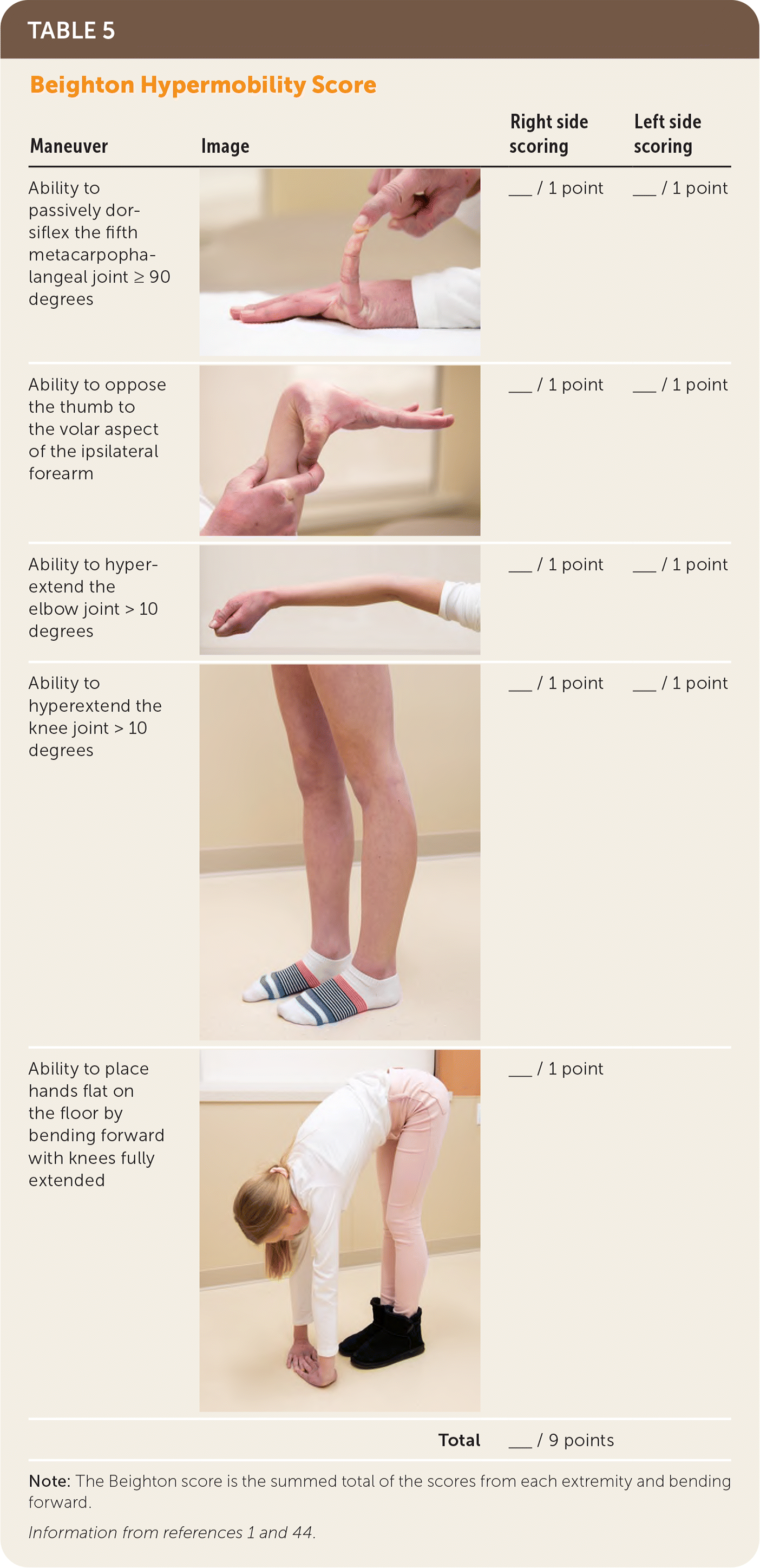
| Maneuver | Image | Right side scoring | Left side scoring |
|---|---|---|---|
| Ability to passively dorsiflex the fifth metacarpophalangeal joint ≥ 90 degrees | ___ / 1 point | ___ / 1 point | |
| Ability to oppose the thumb to the volar aspect of the ipsilateral forearm | ___ / 1 point | ___ / 1 point | |
| Ability to hyperextend the elbow joint > 10 degrees | ___ / 1 point | ___ / 1 point | |
| Ability to hyperextend the knee joint > 10 degrees | ___ / 1 point | ___ / 1 point | |
| Ability to place hands flat on the floor by bending forward with knees fully extended | ___ / 1 point | ||
| Total | ___ / 9 points |
If generalized joint hypermobility is confirmed in patients with suspected hypermobile EDS, the remainder of the hypermobile EDS criteria are sought1 (Figure 26). This involves asking the patient about a history of musculoskeletal symptoms, abdominal hernias, and organ and mitral valve prolapse; examining the skin; testing for arachnodactyly; and measuring the ratio of arm span to height. Figure 1 shows a typical atrophic scar.
DIAGNOSTIC TESTING
No confirmatory test exists, so hypermobile EDS and hypermobility spectrum disorders remain clinical diagnoses.2 Laboratory testing and radiography to evaluate for acquired connective tissue disorder or suspected bone or joint injury are guided by clinical history and physical examination. The presence of marfanoid features requires distinguishing between hypermobile EDS and Marfan-related syndromes. Table 4 lists features that can help to distinguish between these conditions.1,4,21,42 Screening echocardiography should be performed to evaluate for aortic root dilation or mitral valve prolapse in patients with possible hypermobile EDS. Specific genetic testing should be performed for other EDS variants, Marfan and Loeys-Dietz syndromes, and other genetic conditions when suspected (Table 4).1,4,21,42 It often takes several visits to complete a diagnostic evaluation. Many patients who do not meet hypermobile EDS criteria and do not have clear evidence for another specific syndrome will meet criteria for hypermobility spectrum disorders. If the diagnosis remains unclear, referral to a genetics specialist for further evaluation may be required. eTable A lists resources supporting the diagnosis and management of hypermobility syndromes.
| The Ehlers-Danlos Society |
| Diagnostic criteria for hypermobile Ehlers-Danlos syndrome (fillable PDF) |
| https://www.ehlers-danlos.com/wp-content/uploads/hEDS-Dx-Criteria-checklist-1-Fillable-form.pdf |
| Patient education videos |
| https://www.youtube.com/channel/UC652wu-mvi2ghwQN-is7LIQ/videos |
| Professional resources |
| https://www.ehlers-danlos.com/medical-professionals/ |
| Video demonstrating Beighton Scoring System |
| https://www.ehlers-danlos.com/assessing-joint-hypermobility/ |
| Hypermobility Syndromes Association |
| Hypermobility Disorders: An Update for Clinicians |
| https://www.hypermobility.org/hypermobility-disorders-an-update-for-clinicians |
| Royal College of General Practitioners |
| The Ehlers-Danlos Syndromes Toolkit (practical information) |
| https://www.rcgp.org.uk/clinical-and-research/resources/toolkits/ehlers-danlos-syndromes-toolkit.aspx |
Management
The central goals of therapy are managing symptoms, preventing joint injury, and teaching patients about their condition.45–48 Based on limited evidence and expert opinion, the mainstays of management for hypermobile EDS and hypermobility spectrum disorders include patient education, physical and occupational therapy, psychological support, and self-management. Symptoms of hypermobility spectrum disorders may resolve with therapy, persist, or progress to hypermobile EDS. Hypermobile EDS is managed as a lifelong condition because no curative treatments currently exist.
Treatment strategies are diverse because of the many different systems that may be involved. Patients with hypermobile EDS can benefit from a multidisciplinary team that includes physicians, nursing staff, physical therapists, occupational therapists, orthotists, nutritionists and/or lifestyle coaches, psychologists, and community and online support. Specialty care can help in the management of skin, joint, cardiovascular, and gastrointestinal complications and chronic pain. Family physicians play a key role in overseeing and coordinating the complex care that many patients with hypermobile EDS require.
Management of musculoskeletal complaints includes conservative treatments such as physical activity, acetaminophen and nonsteroidal anti-inflammatory drugs, heat and/or cold application, improved ergonomics and posture, relaxation techniques, massage, hydrotherapy, and joint stabilization techniques with bracing and/or taping.47,49,50 Medications that diminish platelet function should generally be avoided in patients with hypermobile EDS who have easy bruising. Physical therapists should customize their education on strengthening exercises, proprioceptive exercises, and joint protection.51 Occupational therapists can help strengthen upper extremity and hand muscles, improve activities of daily living, and introduce patients to adaptive writing instruments and other adaptive tools. Tai chi has shown benefit in patients with osteoarthritis, fibromyalgia, and low back pain,52 although it has not been studied in those who have hypermobile EDS/hypermobility spectrum disorders. The use of splints and orthotics can help selected patients.
Educating patients about lifestyle modifications, management options, and expectations is one of the most important interventions. Encouraging the optimization of sleep, joint protection through the proper amount of regular physical exercise (low impact and low resistance), weight control, avoidance of substance use (e.g., alcohol, nicotine), and the consumption of a healthy diet can decrease pain, injuries, and fatigue and support mobility and functionality. Orthostatic intolerance can be lessened by increasing fluids, increasing salt intake, and using compression stockings.
Qualitative studies describe the experience of patients who have pain and disability with hypermobile EDS that is frequently minimized or invalidated in their social circles, and even in medical settings, because the patients are perceived as looking “normal” or the condition is not recognized.53–55 The complexity of hypermobile EDS/hypermobility spectrum disorders and the lack of familiarity physicians may have can lead them to ignore or be skeptical of patients' experiences with their EDS conditions, possibly having lasting negative impacts on patients.56 Qualitative data strongly support the notion that earlier diagnosis and empathetic, knowledgeable clinical care are highly desired by patients,53–55 but further research is needed to confirm whether earlier diagnosis and skilled, caring support improve outcomes beyond patient satisfaction. The risks and benefits of invasive testing and procedures must be reviewed carefully in patients with all forms of EDS because bleeding complications, inadequate response to regional and local anesthesia, and iatrogenic injury are common.57
Finally, physicians should be aware of and empathetic toward the cognitive deficits, negative emotions, and alterations in activity that can complicate this challenging condition.37 More research is needed, but three small studies of a multidisciplinary approach that includes physical, occupational, and cognitive behavior therapy have shown reduced anxiety, depression, catastrophizing, and kinesiophobia (fear of pain due to movement), with improved physical function and self-efficacy in treated patients.32,58,59 A similar multidisciplinary intervention that lacked cognitive behavior therapy showed no benefit.45
Prognosis
A three-phase natural history of hypermobile EDS has been proposed based on a large Italian case series.49 In this series, patients progressed from generalized joint hypermobility alone with or without joint pain in childhood to having musculoskeletal pain, falls, mixed headache, and functional gastrointestinal disorders by the second and third decades of life. By the third to fourth decades of life, patients developed inflexibility, widespread pain, and limiting fatigue. The prognosis of hypermobile EDS/hypermobility spectrum disorders varies widely and is difficult to predict for individual patients. A convenience sample of children diagnosed with a precursor to hypermobile EDS at a tertiary hospital who were followed for three years found four factors that predicted disease severity and modestly predicted development of disability over time: multisystem involvement, pain, fatigue, and postural control.60 However, variable outcomes were the rule. In adults, chronic pain, gastrointestinal and genitourinary problems, fatigue, restricted mobility, and frequent injuries were most often associated with the functional outcomes of decreased perceived quality of life and decreased participation in activities of daily living.30,53,61
Data Sources: Search in MEDLINE and CINAHL for joint hypermobility (in general, including EDS) and epidemiolog$.mp, risk factors, (pathogenesis or pathogenetic or pathogenic$ or pathogeny).mp, clinical presentation, symptoms, diagnosis, diagnostic criteria, clinical management or prognosis limiting to English and the past 10 years with additional review of bibliographies for relevant articles. American Family Physician editors identified no relevant evidence from POEMs or the Cochrane database. No relevant guidelines were found in the ECRI Guidelines Trust or the U.S. Preventive Services Task Force. Search dates: November 25 and 26, 2019; and October 28, 2020.
The authors thank our medical librarians at Gundersen Health System–La Crosse campus and Drs. Karyn Laursen and Kerry Jedele for their reviews and assistance with the manuscript.
The views expressed in this article are those of the authors and do not necessarily reflect the official position of the Department of Defense or the U.S. government.
