
Am Fam Physician. 2003;68(10):2001-2009
A more recent article on primary immunodeficiency disease in children is available.
Primary immunodeficiencies include a variety of disorders that render patients more susceptible to infections. If left untreated, these infections may be fatal. The disorders constitute a spectrum of more than 80 innate defects in the body's immune system. Primary immunodeficiencies generally are considered to be relatively uncommon. There may be as many as 500,000 cases in the United States, of which about 50,000 cases are diagnosed each year. Common primary immunodeficiencies include disorders of humoral immunity (affecting B-cell differentiation or antibody production), T-cell defects and combined B- and T-cell defects, phagocytic disorders, and complement deficiencies. Major indications of these disorders include multiple infections despite aggressive treatment, infections with unusual or opportunistic organisms, failure to thrive or poor growth, and a positive family history. Early recognition and diagnosis can alter the course of primary immunodeficiencies significantly and have a positive effect on patient outcome.
Currently, more than 80 primary immunodeficiencies are recognized by the World Health Organization.1 While most of these disorders present in childhood, they can manifest later in life. Some primary immunodeficiencies, such as common variable immunodeficiency disorder, present in patients who are in their 20s or 30s. Patients with primary immunodeficiency disorders are susceptible to infections that, if left untreated, may be fatal.
The incidence of most primary immunodeficiencies is uncertain because of the lack of a national registry or reporting by government health surveys. In the United States, as many as 500,000 persons have one of the more than 80 primary immunodeficiencies,2 with about 50,000 cases diagnosed each year.3 The primary immunodeficiencies appear to affect males and females about equally. In a survey of more than 2,700 patients conducted by the Immune Deficiency Foundation,3 48 percent of affected patients were male, and 52 percent were female.
Primary immunodeficiencies can be divided into subgroups based on the component of the immune system that is affected. This article reviews the characteristics of some of the more common primary immunodeficiencies and provides an approach to the initial evaluation of patients suspected of having these disorders.
Background
The body's immune response is made up of a diverse network of defenses, including physical barriers, cellular components, and soluble mediators. The normal immune system has two “arms”: first, it mounts rapid, nonspecific responses (innate immune responses) to initial infection; later, it mounts adaptive immune responses specific to a particular pathogen. Together, these arms work to maintain normal host function and resistance to infection. Disruption of any part of the orchestrated immune response can result in an inability to control infection and subsequent illness.
The innate immune response involves three major cell types: phagocytic cells, such as neutrophils and macrophages; natural killer cells, which have the ability to lyse foreign cells; and antigen-presenting cells, which are involved in the induction of an adaptive immune response. Complement proteins are an important class of soluble mediators of the innate immune response and serve to promote inflammation and microbial killing of extra-cellular pathogens.
The adaptive immune system includes T and B lymphocytes and can be divided into cellular and humoral responses. The cellular immune response is mediated primarily by T cells and limits intracellular infections by organisms such as viruses, parasites, and mycobacteria. Antibodies, the key feature of the humoral response, are produced by activated B cells to help control the spread of extracellular pathogens. T-lymphocyte and B-lymphocyte responses are not independent of one another; for example, B cells can activate antigen-specific T cells for a cellular immune response, while an efficient B-cell antibody response depends in part on T-cell activation of B lymphocytes. Thus, defects in either cell type have the potential to affect both cellular and humoral immunity to varying degrees.
Characteristics of Primary Immunodeficiencies
| Subgroup | Onset | Pattern of infection | Other features |
|---|---|---|---|
| Disorders of humoral immunity (B-cell differentiation and antibody production) | After 6 months of age; can present in adulthood | Encapsulated bacteria:Haemophilus influenzae, pneumococci, streptococci Fungi and parasites:Giardia lamblia, Cryptosporidium species Virus: enterovirus (especially with X-linked agammaglobulinemia) | Recurrent infections: sinus infections, otitis media, bronchiectasis |
| Chronic gastrointestinal tract problems, including malabsorption | |||
| Autoimmunity | |||
| Postvaccination paralytic polio (with live oral poliovirus vaccine) | |||
| T-cell and mixed disorders (combined B-cell and T-cell defects) | Before 6 months of age | Various opportunistic infections: Mycobacterium species, cytomegalovirus, Epstein-Barr virus, varicella virus, enterovirus, Candida species,Pneumocystis carinii (pneumonia) | Failure to thrive |
| Oral thrush | |||
| Graft-versus-host disease from maternal lymphocytes | |||
| Excess diarrhea | |||
| Postvaccination disseminated bacille Calmette-Guerin infection or paralytic polio | |||
| Phagocytic disorders | Infancy or childhood | Bacteria:Staphylococcus aureus, Pseudomonas species, Serratia species Klebsiella species Fungi and parasites: Candida species, Nocardia species, Aspergillus species | Unusually severe infections by common pathogens |
| Granuloma formation, including granulomatous enteritis | |||
| Poor wound healing | |||
| Abscesses, skin infections | |||
| Oral cavity infections | |||
| Anorectal infections | |||
| Complement disorders | Any age | Neisseria infections, including meningococcal and gonococcal infections | Rheumatoid disorders |
| Lupus-like syndrome | |||
| Scleroderma |
| Disorders (percentage of all primary immunodeficiencies) | Genetic inheritance pattern | Incidence, if known | Sex affected | Age at diagnosis | ||
|---|---|---|---|---|---|---|
| Disorders of humoral immunity: B-cell differentiation and antibody production (~ 50) | ||||||
| Common variable immunodeficiency | Undetermined | One case per 10,000 to 50,000 persons | Both | >2 years; can be in 20s or 30s | ||
| Selective IgA deficiency | Undetermined | About one case per 300 to 700 persons | Both | >4 years | ||
| Bruton's or X-linked agammaglobulinemia | X-linked | Undetermined | Males | >6 months | ||
| T-cell defects and combined B-cell and T-cell defects (~30) | ||||||
| Severe combined immunodeficiency | X-linked | One case per 100,000 to 500,000 persons | Males | <6 months | ||
| T-cell deficient, B-cell competent | Autosomal recessive | Both | <6 months | |||
| T-cell deficient, B-cell deficient | Autosomal recessive | Both | <6 months | |||
| DiGeorge syndrome | Autosomal dominant or spontaneous | Undetermined | Both | <6 months | ||
| Wiskott-Aldrich syndrome | X-linked | Undetermined | Males | <6 months | ||
| Ataxia-telangiectasia | Autosomal recessive | Undetermined | Both | >5 years | ||
| X-linked hyper IgM | X-linked | Undetermined | Males | Variable | ||
| Phagocytic disorders (~ 18) | ||||||
| Chronic granulomatous disease | X-linked (70% of cases) or autosomal recessive (22% of cases) | One case per 200,000 persons | Males > females | Usually <5 years; diagnosis can be in 20s and 30s | ||
| Complement disorders (~ 2) | ||||||
| Complement deficiencies (at least 16 distinct disorders) | Autosomal recessive, autosomal dominant, or X-linked | Undetermined | Both | Any age | ||
DISORDERS OF HUMORAL IMMUNITY
Disorders of humoral immunity affect B-cell differentiation and antibody production. Collectively, these disorders account for approximately 50 percent of primary immunodeficiencies.5
Patients with antibody deficiencies often present after six months of age, when maternal antibodies are lost, but they can present in adulthood.10 Typically, these patients develop infections with encapsulated bacteria. Recurrent bacterial sinus and pulmonary infections are the hallmark of antibody primary immunodeficiencies. Patients with humoral primary immunodeficiencies have an intact cellular immune system; thus, they are able to handle most viral and fungal pathogens, a factor that can help to distinguish these disorders clinically.
In the United States, common variable immunodeficiency is the most frequently diagnosed primary immunodeficiency.3 The term “common variable immunodeficiency” encompasses a heterogeneous group of disorders that cause hypogammaglobulinemia (serum IgA levels below 5 mg per dL [0.05 g per L]).1,11 Onset can occur after two years of age, but the average age of onset is the middle to late 20s.10 Patients with common variable immunodeficiency have a poor response to vaccines (decreased IgG antibody response) and an increased risk of developing auto-immune disorders and malignancy.
Of the primary immunodeficiency disorders, selective IgA deficiency may have the highest incidence (one case per 300 to 700 persons, according to estimates based on blood donation analyses), but the disorder is often asymptomatic and undiagnosed.3,12 Patients with symptoms often have sinusitis and respiratory tract infections, along with gastrointestinal involvement. All patients with IgA deficiency are at increased risk for allergies and autoimmune diseases. Although serum IgA levels are below 5 mg per dL, serum IgG and IgM levels are in the normal range. In contrast to patients with common variable immunodeficiency, patients with IgA deficiency have a normal IgG response to vaccinations.
Bruton's or X-linked agammaglobulinemia is caused by mutation or absence of the Bruton's tyrosine kinase gene.13 Early B-cell development is arrested, and serum immunoglobulins (IgG, IgA, IgM) are markedly deficient or totally absent.10 Onset of recurrent bacterial infections is usually at the end of the first year of life; however, patients with the disorder may not present until the age of three to five years.
T-CELL DEFECTS AND COMBINED B-CELL AND T-CELL DEFECTS
Disruption of the cellular immune response is observed in patients with defects in T cells or both T and B cells. These primary immunodeficiency disorders are generally more severe than antibody deficiencies. Affected patients often present early in life with failure to thrive and disseminated infection.7 DiGeorge syndrome is one of the most recognized disorders in this category, and severe combined immunodeficiency is the most severe. General features of this class of diseases include overwhelming viral and fungal infections.
DiGeorge syndrome results in abnormal migration of the third and fourth branchial pouches during embryogenesis, with hypoplasia to aplasia of the thymus and parathyroid glands. The syndrome most often is caused by a deletion in chromosome 22q11. Associated defects include truncal cardiac malformations (e.g., truncus arteriosis, Fallot's tetralogy) and dysmorphic facial features. Other diagnostic criteria include a reduced CD3+ T-cell count (less than 500 per mm3 [0.5 × 109 per L]) and hypocalcemia of greater than three weeks' duration.11 [Evidence level C: consensus/expert opinion]
Severe combined immunodeficiency is associated with profound deficiencies of T-cell and B-cell function (and sometimes natural killer cell function). This disorder is characterized by severe opportunistic infections, or by chronic diarrhea and failure to thrive in infancy. Laboratory findings typically demonstrate severe lymphopenia. About one half of cases are X-linked, and one half are autosomal recessive.14 Infants with this primary immunodeficiency disorder are at risk for graft-versus-host disease because they lack the ability to reject foreign tissue, such as maternal T cells that cross into the fetal circulation in utero.
Wiskott-Aldrich syndrome is an X-linked recessive syndrome characterized by thrombocytopenia, small platelets and platelet dysfunction, eczema, and susceptibility to infections.7 Infants typically present with prolonged bleeding from the circumcision site, bloody diarrhea, or excessive bruising. Patients with this primary immunodeficiency disorder are at risk for autoimmune diseases and cancer.
Ataxia-telangiectasia (Louis-Bar's syndrome) is a progressive neurologic disorder associated with cerebellar ataxia, oculocutaneous telangiectasias, chronic respiratory infections, a high incidence of malignancy, and variable humoral and cellular immunodeficiency. Patients with this disorder have difficulty walking and generally are wheelchair-bound by the teenage years.
PHAGOCYTIC DISORDER: CHRONIC GRANULOMATOUS DISEASE
Chronic granulomatous disease, the most frequently diagnosed phagocytic primary immunodeficiency, is more common in males than in females. In this disease, deficiency of nicotinamide adenine dinucleotide phosphate oxidase in phagocytes results in defective elimination of extracellular pathogens such as bacteria and fungi. Patients with chronic granulomatous disease are more susceptible to infection with catalase-positive organisms (e.g., staphylococci) that require phagocytic activity for clearance. Aspergillus infection is the most common cause of death in patients with phagocytic primary immunodeficiency disorders.4
COMPLEMENT DEFICIENCIES
Complement disorders account for only 2 percent of all primary immunodeficiency disorders.6 They result from the disruption of one of the proteins involved in the classic or nonclassic activation pathways of the complement response.15 Defects in the classic pathway account for the more common type of complement deficiency, and patients often have a high number of autoimmunity disorders, including lupus-like syndromes. Patients with defects of the alternative pathway characteristically present with Neisseria infection.15
Diagnosis of Primary mmunodeficiencies
WARNING SIGNS AND SYMPTOMS
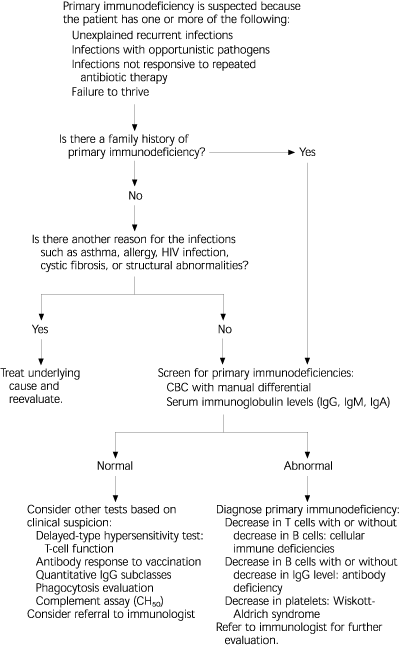
The National Institute of Child Health and Human Development recently initiated an educational program to raise awareness of primary immunodeficiencies. As a part of this program, the Jeffrey Modell Foundation developed a list of warning signs for primary immunodeficiency.2 These warning signs, along with other common presenting signs, are listed in Table 3.2,6,16 A general approach to the evaluation of patients with suspected primary immunodeficiency is presented in Figure 1.
| Medical history |
| Eight or more ear infections in one year |
| Two or more serious sinus infections inone year |
| Two or more bouts of pneumonia in oneyear |
| Two or more deep-seated infections, orinfections in unusual areas |
| Recurrent deep skin or organabscesses |
| Need for intravenous antibiotic therapyto clear infection |
| Infections with unusual or opportunisticorganisms |
| Family history of primary immunodeficiency |
| Physical signs |
| Poor growth, failure to thrive |
| Absent lymph nodes or tonsils |
| Skin lesions: telangiectasias, petechiae, dermatomyositis, lupus-like rash |
| Ataxia (with ataxia-telangiectasia) |
| Oral thrush after one year of age |
| Oral ulcers |
LABORATORY TESTING
When primary immunodeficiency is suspected, initial laboratory studies include a complete blood cell count (CBC) with manual differential, quantitative immunoglobulin measurements (IgG, IgM, IgA), measurements of functional antibodies against immunized antigens, and delayed-type hypersensitivity skin tests (Table 4).6,16,17 The CBC with manual differential can detect deficiencies in immune cells and platelets. In most instances, a normal CBC eliminates the diagnosis of T-cell defects or combined B-cell and T-cell defects.
| Laboratory test | Screens for… | What to look for… |
|---|---|---|
| Complete blood cell count with manual differential | T-cell, B-cell, and mixed B-cell and T-cell defects | Decreased numbers of T cells, B cells, or platelets |
| Delayed-type hypersensitivity skin test | T-cell defects | Negative result signaling possible impaired T-cell response* |
| Serum IgG, IgM, and IgA levels | Humoral immunodeficiencies | Decrease in any or all of the serum immunoglobulins |
| Antibody testing to specific antigens after vaccination | Humoral immunodeficiencies | Decreased or absent antibody response to vaccination † |
| Total hemolytic complement assay (CH50) | Complement deficiencies | Decreased or absent proteins if there is a deficiency in the classic pathway |
| Nitroblue tetrazolium test | Phagocytic disorders | Abnormal test result ‡ |
Caution should be used when assessing immunologic function in newborns. Because of engrafted maternal immune cells, neonates may have both a falsely elevated lymphocyte count and evidence of graft-versus-host disease.18 If severe combined immunodeficiency is strongly suspected and the lymphocyte count is normal or nearly normal, further investigation is warranted to determine the origin of the immune cells.
When a diagnosis is uncertain, additional tests, such as genetic assays or immunophenotyping, might be performed in consultation with a pediatric immunologist.1
Management of Patients with Primary Immunodeficiencies
INTRAVENOUS IMMUNE GLOBULIN
For the past 20 years, intravenously administered immune globulin (IVIG) has been used in the treatment of agammaglob-ulinemia.19 This agent is now standard therapy for most antibody deficiencies. Most commonly, IVIG is used in patients with X-linked agammaglobulinemia, common variable immunodeficiency, X-linked hyper IgM, severe combined immunodeficiency, Wiskott-Aldrich syndrome, and selective IgG class deficiency.3,19–21
IVIG also is used, or is being considered for use, in a wide variety of other illnesses. Consequently, its limited availability is a concern.21
BONE MARROW TRANSPLANTATION
Bone marrow transplants from HLA-identical donors can be curative in patients with cellular immune deficiencies such as severe combined immunodeficiency, Wiskott-Aldrich syndrome, and DiGeorge syndrome, and may be beneficial in patients with chronic granulomatous disease.4,14 Bone marrow transplantation currently has no role in the treatment of antibody deficiencies.9
HLA-identical donors are not always available. Long-term survival may be lower with bone marrow transplants from haploidentical donors. Thus, investigations of alternative strategies, such as gene therapy, could benefit the management of patients with primary immunodeficiency disorders who otherwise would require bone marrow transplantation.
ANTIBIOTICS AND OTHER THERAPIES
When recurrent infections are a problem, many patients with primary immunodeficiencies are managed with antibiotics alone or in combination with IVIG. For example, in patients with chronic granulomatous disease, prophylactic therapy with trimethoprim-sulfamethoxazole (Bactrim, Septra) reduces the incidence of severe infections by 50 percent.4 Similarly, treatment for complement deficiencies is directed at preventing infection, and consists of antibiotic prophylaxis and immunizations for encapsulated bacteria (e.g., heptovalent pneumococcal vaccine, Haemophilus b conjugate vaccine, meningococcal polysaccharide vaccine).14
Other treatments for primary immunodeficiencies include enzyme replacement in patients with adenosine deaminase deficiency (a subtype of severe combined immunodeficiency) and cytokine therapy in patients with chronic granulomatous disease.8
Vaccines and Blood Products: Cautions and Contraindications
Most patients with primary immunodeficiencies should not receive live virus vaccines, including live oral poliovirus vaccine (OPV). Because of the risk of infection, OPV also should not be given to persons in close contact with these patients.14 In addition, most patients with primary immunodeficiencies should not receive measles, bacille Calmette-Guérin, and varicella vaccines. One exception would be patients with B-cell deficiency, who should receive varicella vaccine.
Patients with T-cell deficiencies should receive cytomegalovirus-negative irradiated blood products because of the risk of infection and graft-versus-host disease from lymphocytes in the donor blood. Patients with IgA deficiency need to be informed about the possibility of having a serious reaction to plasma or blood transfusions, because of antibodies to IgA.5