
Am Fam Physician. 2024;109(5):388-390
Author disclosure: No relevant financial relationships.
The world of cystic fibrosis (CF) is changing. Patients are living longer, healthier lives and are now facing many of the same ongoing health challenges as people without CF. Due to significant advances in diagnostic and therapeutic options, the average age of survival for people with the condition in the United States was approximately 53 years in 2021 compared with 36 years in 2006.1–3 Approximately 40,000 people in the United States are living with CF. The disease is found in patients of all racial and ethnic groups. Currently, it is most common in White people (91.2%), followed by Black (3.5%) and Asian (0.5%) people. Additionally, 10% of patients are Hispanic.3 Lower rates of prevalence in minority groups may be due to diagnostic bias; physicians should take care to consider each CF diagnosis based on clinical presentation and not demographics.
Beginning in 2010, newborn screening for CF was universally mandated in the United States; however, there is significant variability in testing. The diagnosis of CF in patients who are racial minorities or who have mild CF or rare mutations may be missed.4 As diagnostic effectiveness improves, many racial and ethnic minorities with CF are being identified worldwide.5 However, these patients tend to be diagnosed later in life and have poorer outcomes.4,6,7 Because access to therapies is key to improving quality of life and longevity, CF should be diagnosed expeditiously.8,9 Regardless of the race and ethnicity of the patient, newborn screening results, or perinatal carrier testing results, it is essential to include CF in the differential diagnosis of unexplained poor weight gain, poor growth, or chronic respiratory or gastrointestinal symptoms (Figure 1).
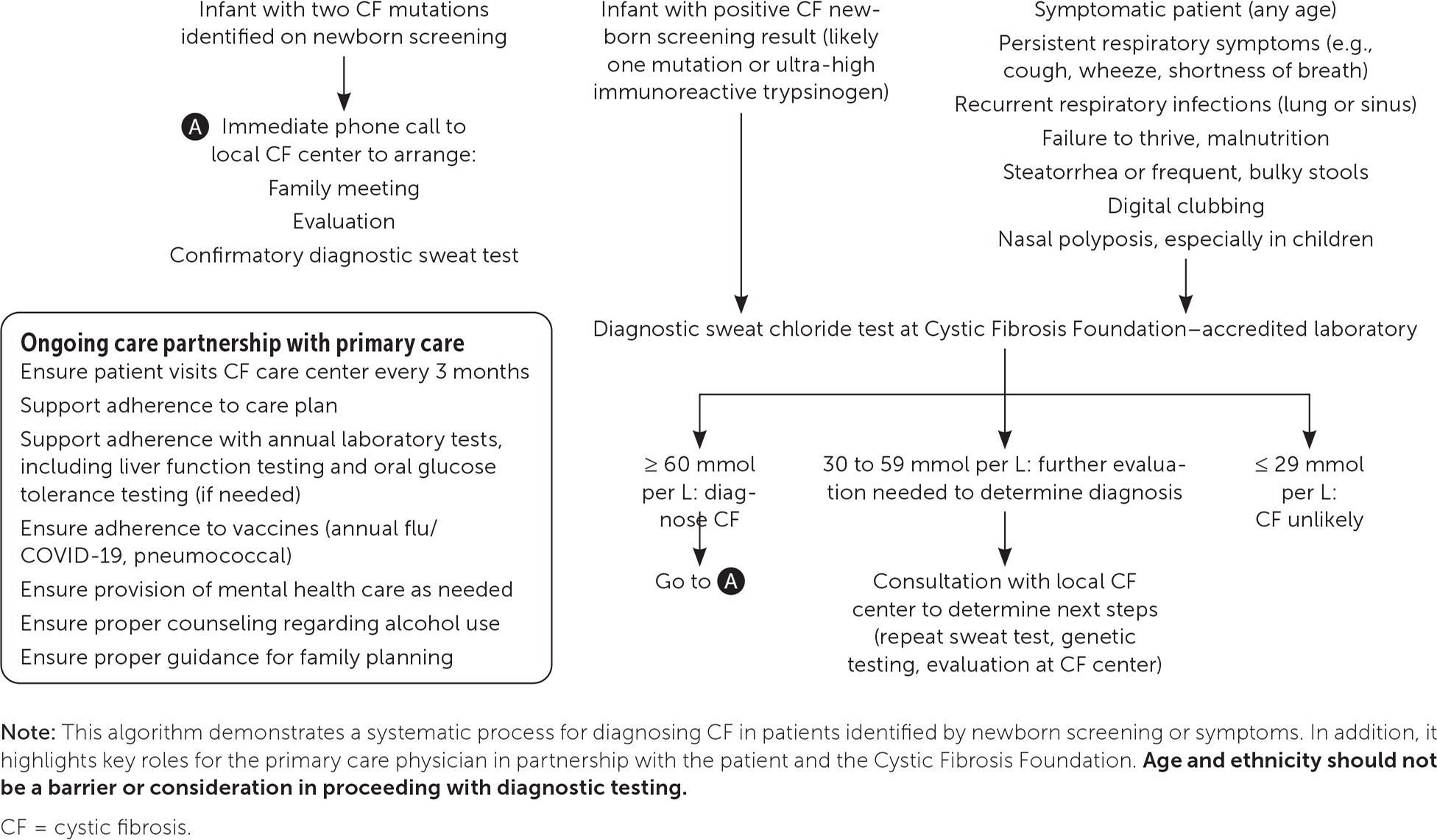
CF is an autosomal recessive disease that results in a mutation on chromosome 7.9 This mutation is in the CF transmembrane conductance regulator protein, which is a chloride channel on the apical membrane of cells that regulates chloride flow and transport of other molecules. Mutations can be grouped into five classes based on their functional defects.10 Because the CF transmembrane conductance regulator protein affects a variety of cell types, mutations can cause disease in multiple organ systems, most commonly the respiratory (including the upper airway), gastrointestinal, endocrine (including the pancreas), and reproductive systems.8 There are more than 2,000 known mutations for CF with many beyond the scope of newborn screening or prenatal carrier testing programs.11 This further supports the importance of diagnostic sweat testing in patients with symptoms, performed at a laboratory and care center accredited by the Cystic Fibrosis Foundation (https://apps.cff.org/ccd).
Like any chronic condition, CF requires attention to preventive care, lifestyle modifications, disease and drug complications, drug-drug interactions, and mental health needs. A list of commonly used CF medications is provided in eTable A. In addition to guideline-directed therapeutic and diagnostic care, the Cystic Fibrosis Foundation recommends that patients receive multidisciplinary care from CF care centers every 3 months.12 Unfortunately, some patients may not be using a CF care center or be aware of new therapeutics, and some may find care center recommendations difficult to follow.13 Family physicians can reinforce guidelines and patient adherence to these recommendations, identify and manage complications, promote routine health maintenance and vaccinations, and assist with the transition from childhood to adult CF care.
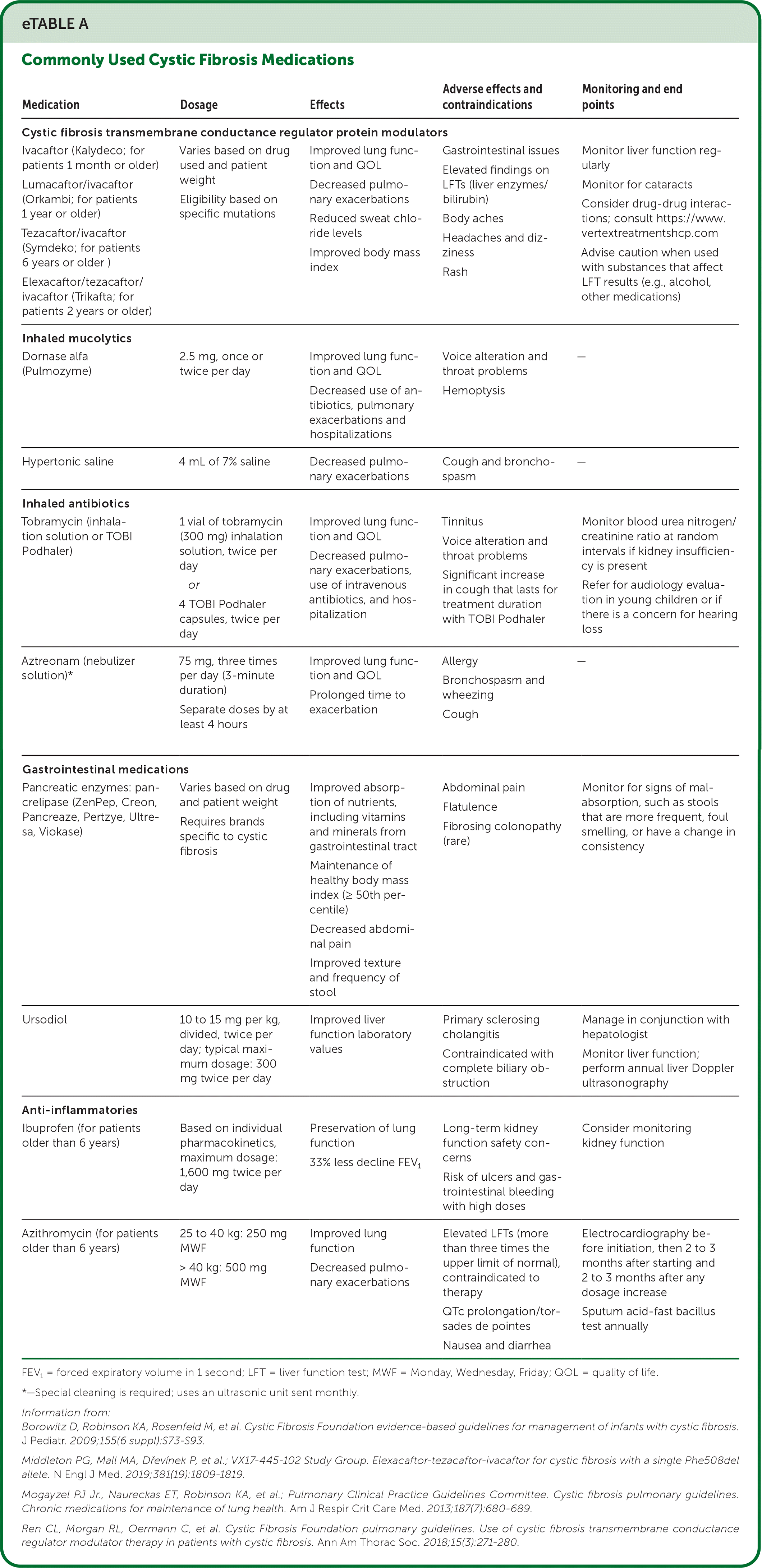
| Medication | Dosage | Effects | Adverse effects and contraindications | Monitoring and end points |
|---|---|---|---|---|
| Cystic fibrosis transmembrane conductance regulator protein modulators | ||||
| Ivacaftor (Kalydeco; for patients 1 month or older) Lumacaftor/ivacaftor (Orkambi; for patients 1 year or older) Tezacaftor/ivacaftor (Symdeko; for patients 6 years or older) Elexacaftor/tezacaftor/ivacaftor (Trikafta; for patients 2 years or older) | Varies based on drug used and patient weight Eligibility based on specific mutations | Improved lung function and QOL Decreased pulmonary exacerbations Reduced sweat chloride levels Improved body mass index | Gastrointestinal issues Elevated findings on LFTs (liver enzymes/bilirubin) Body aches Headaches and dizziness Rash | Monitor liver function regularly Monitor for cataracts Consider drug-drug interactions; consult https://www.vertextreatmentshcp.com Advise caution when used with substances that affect LFT results (e.g., alcohol, other medications) |
| Inhaled mucolytics | ||||
| Dornase alfa (Pulmozyme) | 2.5 mg, once or twice per day | Improved lung function and QOL Decreased use of antibiotics, pulmonary exacerbations and hospitalizations | Voice alteration and throat problems Hemoptysis | — |
| Hypertonic saline | 4 mL of 7% saline | Decreased pulmonary exacerbations | Cough and bronchospasm | — |
| Inhaled antibiotics | ||||
| Tobramycin (inhalation solution or TOBI Podhaler) | 1 vial of tobramycin (300 mg) inhalation solution, twice per day or 4 TOBI Podhaler capsules, twice per day | Improved lung function and QOL Decreased pulmonary exacerbations, use of intravenous antibiotics, and hospitalization | Tinnitus Voice alteration and throat problems Significant increase in cough that lasts for treatment duration with TOBI Podhaler | Monitor blood urea nitrogen/creatinine ratio at random intervals if kidney insufficiency is present Refer for audiology evaluation in young children or if there is a concern for hearing loss |
| Aztreonam (nebulizer solution)* | 75 mg, three times per day (3-minute duration) Separate doses by at least 4 hours | Improved lung function and QOL Prolonged time to exacerbation | Allergy Bronchospasm and wheezing Cough | — |
| Gastrointestinal medications | ||||
| Pancreatic enzymes: pancrelipase (ZenPep, Creon, Pancreaze, Pertzye, Ultresa, Viokase) | Varies based on drug and patient weight Requires brands specific to cystic fibrosis | Improved absorption of nutrients, including vitamins and minerals from gastrointestinal tract Maintenance of healthy body mass index (≥ 50th percentile) Decreased abdominal pain Improved texture and frequency of stool | Abdominal pain Flatulence Fibrosing colonopathy (rare) | Monitor for signs of malabsorption, such as stools that are more frequent, foul smelling, or have a change in consistency |
| Ursodiol | 10 to 15 mg per kg, divided, twice per day; typical maximum dosage: 300 mg twice per day | Improved liver function laboratory values | Primary sclerosing cholangitis Contraindicated with complete biliary obstruction | Manage in conjunction with hepatologist Monitor liver function; perform annual liver Doppler ultrasonography |
| Anti-inflammatories | ||||
| Ibuprofen (for patients older than 6 years) | Based on individual pharmacokinetics, maximum dosage: 1,600 mg twice per day | Preservation of lung function 33% less decline FEV1 | Long-term kidney function safety concerns Risk of ulcers and gastrointestinal bleeding with high doses | Consider monitoring kidney function |
| Azithromycin (for patients older than 6 years) | 25 to 40 kg: 250 mg MWF > 40 kg: 500 mg MWF | Improved lung function Decreased pulmonary exacerbations | Elevated LFTs (more than three times the upper limit of normal), contraindicated to therapy QTc prolongation/torsades de pointes Nausea and diarrhea | Electrocardiography before initiation, then 2 to 3 months after starting and 2 to 3 months after any dosage increase Sputum acid-fast bacillus test annually |
Lung disease is a major cause of morbidity and mortality, and patients with CF require a regimen of techniques and mucolytic medications for airway clearance.14 Aerobic exercise augments airway clearance, preserves pulmonary function, increases cardiovascular fitness, and enhances quality of life.15,16 Office spirometry should be repeated over time as an indicator of lung function and other complications.
Acute complications of CF include pulmonary exacerbations, which typically result in weight loss, worsening cough and lung function, and a potential increase in supplemental oxygen needs. Depending on severity, patients with such exacerbations can be treated as outpatients with oral antibiotics, but they may require inpatient admission for intravenous antibiotic therapy to treat pulmonary exacerbations. Less common acute complications include pneumothorax and hemoptysis; these require urgent evaluation and treatment at a tertiary care facility.17
The nutritional status of patients with CF determines long-term survival and affects lung function decline. For adults, an ideal dietary plan maintains more than 90% of ideal body weight, or a body mass index of 22 to 25 kg per m2, and includes supplementation with fat-soluble vitamins.16 Because 85% of patients with CF develop pancreatic insufficiency, fecal pancreatic elastase should be tested at diagnosis and then annually. Histamine H2 blockers or proton pump inhibitors may be used to augment pancreatic enzyme effects and treat common symptoms of gastroesophageal reflux disease. Due to high caloric needs, some patients require supplemental gastrostomy tube feeding. Constipation is common and often underdiagnosed due to abnormal stooling caused by pancreatic insufficiency. Patients with CF may also have significant liver disease, and routine monitoring of the entire gastrointestinal tract is required.18,19
Other chronic complications of CF include diabetes mellitus and bone disease. CF-related diabetes occurs in about 30% of adults with CF, and A1C is not a reliable method for diagnosis or determining control; therefore, ongoing glucose monitoring is recommended, with insulin as the primary treatment.14,15 Additionally, up to 12% of patients develop immune-mediated joint disease. Acute episodes present with pain and joints that are red, hot, and swollen. Episodes last 7 to 10 days and may affect any joints, usually asymmetrically. Short courses of nonsteroidal anti-inflammatory drugs and corticosteroids are useful in the management of this recurrent issue.15 eTable B summarizes ongoing health monitoring for patients with CF.
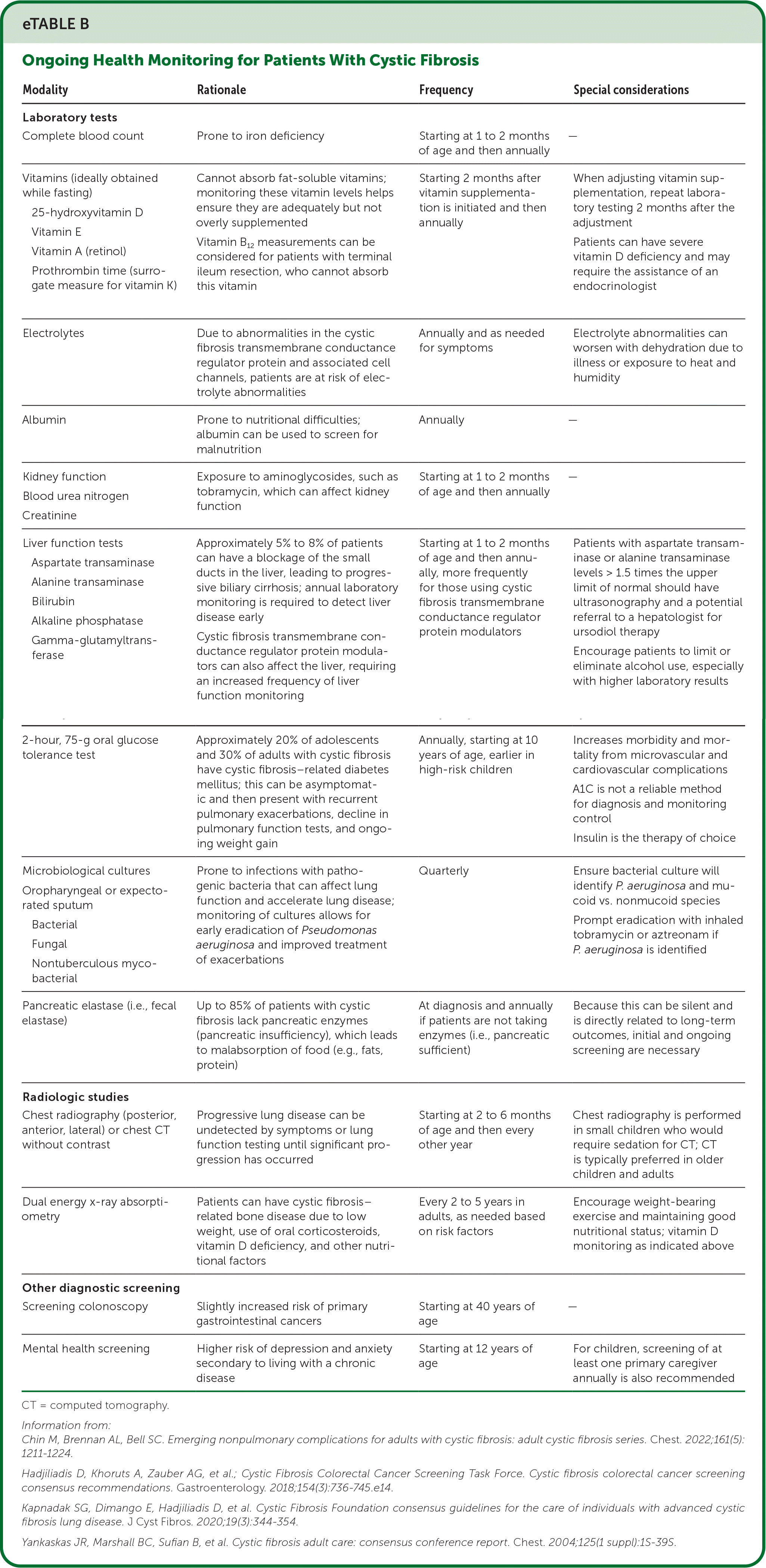
| Modality | Rationale | Frequency | Special considerations |
|---|---|---|---|
| Laboratory tests | |||
| Complete blood count | Prone to iron deficiency | Starting at 1 to 2 months of age and then annually | — |
| Vitamins (ideally obtained while fasting) 25-hydroxyvitamin D Vitamin E Vitamin A (retinol) Prothrombin time (surrogate measure for vitamin K) | Cannot absorb fat-soluble vitamins; monitoring these vitamin levels helps ensure they are adequately but not overly supplemented Vitamin B12 measurements can be considered for patients with terminal ileum resection, who cannot absorb this vitamin | Starting 2 months after vitamin supplementation is initiated and then annually | When adjusting vitamin supplementation, repeat laboratory testing 2 months after the adjustment Patients can have severe vitamin D deficiency and may require the assistance of an endocrinologist |
| Electrolytes | Due to abnormalities in the cystic fibrosis transmembrane conductance regulator protein and associated cell channels, patients are at risk of electrolyte abnormalities | Annually and as needed for symptoms | Electrolyte abnormalities can worsen with dehydration due to illness or exposure to heat and humidity |
| Albumin | Prone to nutritional difficulties; albumin can be used to screen for malnutrition | Annually | — |
| Kidney function Blood urea nitrogen Creatinine | Exposure to aminoglycosides, such as tobramycin, which can affect kidney function | Starting at 1 to 2 months of age and then annually | — |
| Liver function tests Aspartate transaminase Alanine transaminase Bilirubin Alkaline phosphatase Gamma-glutamyltransferase | Approximately 5% to 8% of patients can have a blockage of the small ducts in the liver, leading to progressive biliary cirrhosis; annual laboratory monitoring is required to detect liver disease early Cystic fibrosis transmembrane conductance regulator protein modulators can also affect the liver, requiring an increased frequency of liver function monitoring | Starting at 1 to 2 months of age and then annually, more frequently for those using cystic fibrosis transmembrane conductance regulator protein modulators | Patients with aspartate transaminase or alanine transaminase levels > 1.5 times the upper limit of normal should have ultrasonography and a potential referral to a hepatologist for ursodiol therapy Encourage patients to limit or eliminate alcohol use, especially with higher laboratory results |
| 2-hour, 75-g oral glucose tolerance test | Approximately 20% of adolescents and 30% of adults with cystic fibrosis have cystic fibrosis–related diabetes mellitus; this can be asymptomatic and then present with recurrent pulmonary exacerbations, decline in pulmonary function tests, and ongoing weight gain | Annually, starting at 10 years of age, earlier in high-risk children | Increases morbidity and mortality from microvascular and cardiovascular complications A1C is not a reliable method for diagnosis and monitoring control Insulin is the therapy of choice |
| Microbiological cultures Oropharyngeal or expectorated sputum Bacterial Fungal Nontuberculous mycobacterial | Prone to infections with pathogenic bacteria that can affect lung function and accelerate lung disease; monitoring of cultures allows for early eradication of Pseudomonas aeruginosa and improved treatment of exacerbations | Quarterly | Ensure bacterial culture will identify P. aeruginosa and mucoid vs. nonmucoid species Prompt eradication with inhaled tobramycin or aztreonam if P. aeruginosa is identified |
| Pancreatic elastase (i.e., fecal elastase) | Up to 85% of patients with cystic fibrosis lack pancreatic enzymes (pancreatic insufficiency), which leads to malabsorption of food (e.g., fats, protein) | At diagnosis and annually if patients are not taking enzymes (i.e., pancreatic sufficient) | Because this can be silent and is directly related to long-term outcomes, initial and ongoing screening are necessary |
| Radiologic studies | |||
| Chest radiography (posterior, anterior, lateral) or chest CT without contrast | Progressive lung disease can be undetected by symptoms or lung function testing until significant progression has occurred | Starting at 2 to 6 months of age and then every other year | Chest radiography is performed in small children who would require sedation for CT; CT is typically preferred in older children and adults |
| Dual energy x-ray absorptiometry | Patients can have cystic fibrosis– related bone disease due to low weight, use of oral corticosteroids, vitamin D deficiency, and other nutritional factors | Every 2 to 5 years in adults, as needed based on risk factors | Encourage weight-bearing exercise and maintaining good nutritional status; vitamin D monitoring as indicated above |
| Other diagnostic screening | |||
| Screening colonoscopy | Slightly increased risk of primary gastrointestinal cancers | Starting at 40 years of age | — |
| Mental health screening | Higher risk of depression and anxiety secondary to living with a chronic disease | Starting at 12 years of age | For children, screening of at least one primary caregiver annually is also recommended |
The American College of Obstetricians and Gynecologists recommends prenatal carrier testing for CF, including prenatal patients with CF who may not know their genetic mutations.20 Patients with CF experience issues with fertility, but improvements in quality of life and lung function with CF transmembrane conductance regulator protein modulators have enabled more women with CF to become pregnant.21 Expert guidelines emphasize optimization of prepregnancy health, genetic counseling, and partnership with a CF care center.21 The Cystic Fibrosis Foundation also recommends that a genetic counseling component be a part of newborn screening programs to assist families with understanding results and future family planning.22 Due to a lack of this component in many programs, telemedicine is being explored as a more widely feasible option for genetic counseling for CF.23
Given these advancements, family physicians play a crucial role in caring for patients with CF and connecting them with care centers and the Cystic Fibrosis Foundation website to take advantage of the new treatment and support options awaiting them there. See eTable A for information on commonly used medications and eTable B for monitoring guidance.
