
Am Fam Physician. 2005;72(11):2259-2262
Author disclosure: Nothing to disclose.
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Air Force Medical Service, the U.S. Air Force at large, the National Human Genome Research Institute, or the National Institutes of Health.
The authors thank John M. Graham, Jr., M.D., Sc.D., and Nasreen Malik, M.D., for assistance with the preparation of the manuscript.
To complement the 2005 Annual Clinical Focus on medical genomics, AFP is publishing a series of short reviews on genetic syndromes. This series was designed to increase awareness of these diseases so that family physicians can recognize and diagnose children with these disorders and understand the kind of care they might require in the future. This review discusses Klinefelter syndrome.
Klinefelter syndrome is caused by an additional X chromosome in males (47,XXY). Clinical findings are nonspecific during childhood; thus, the diagnosis commonly is made during adolescence or adulthood in males who have small testes with hypergonadotropic hypogonadism and gynecomastia. Virtually all men with Klinefelter syndrome are infertile.
Epidemiology
Approximately one in 1,000 boys is born with an additional X chromosome—47,XXY, the karyotype that causes Klinefelter syndrome.1 This karyotype is detected at or before birth in 10 percent of affected boys, and it is found during adulthood in 25 percent of affected men.2 Almost all men with a 47,XXY karyotype will be infertile; Klinefelter syndrome accounts for 3 percent of male infertility.3 Klinefelter syndrome is common in infertile men with oligospermia or azoospermia (5 to 10 percent).2
Clinical Presentation
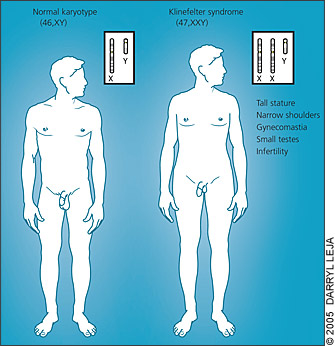
The most overt phenotypic features of Klinefelter syndrome are caused by testosterone deficiency and, directly or indirectly, by unsuppressed follicle-stimulating and luteinizing hormones. Affected men typically have (in decreasing order of frequency): infertility, small testes, decreased facial hair, gynecomastia, decreased pubic hair, and a small penis (Table 1).2 Because of their long legs, men with Klinefelter syndrome often are taller than predicted based on parental height. Body habitus may be feminized (Figure 1). In childhood, when there is a relative quiescence in the hormonal milieu, ascertainment of the syndrome may be difficult because the effects of hypogonadism (i.e., small external genitalia and firm testes) may be subtle or not present at all.
| Infertility (azoospermia or oligospermia) |
| Small, firm testes |
| Hypergonadotropic hypogonadism |
| Gynecomastia |
| Tall, slender body habitus with long legs and shorter torso |
| Osteoporosis (in young or middle-age men) |
| Motor delay or dysfunction |
| Speech and language difficulties |
| Attention deficits |
| Learning disabilities |
| Dyslexia or reading dysfunction |
| Psychosocial or behavioral problems |
Persistent androgen deficiency in adulthood may result in loss of libido, decreased muscle bulk and tone, decreased bone mineral density, a propensity for thromboembolism, and an increased risk of mortality from diabetic and cardiovascular complications.4
Specific cognitive deficits in language comprehension and executive functioning are part of the Klinefelter syndrome phenotype.5 Although most studies describe only mild cognitive impairment (e.g., mild global delay in gross and fine motor development, speech, and language), Klinefelter syndrome has been identified in 0.4 percent of boys with special education needs and no known diagnosis, and in 1.2 percent of boys with mental retardation of unknown etiology. In these cases, the boys initially were referred for consideration of fragile X syndrome.6,7 Of note, the 0.4 percent of boys with Klinefelter syndrome in the special-education cohort is likely to represent less than half the actual prevalence, because only paternally acquired disease was identified.
Diagnosis
Because most boys with Klinefelter syndrome appear similar to boys with normal karyotypes, the disorder typically is identified in adulthood, when infertility or gynecomastia are common presentations. However, by this time, a therapeutic window during adolescence for treatment of hypogonadism, cognitive impairments, and psychosocial factors has been missed. Late or incomplete puberty should prompt further examination. Testicular size can be measured with a Prader orchidometer or ultrasonography. Middle school–age boys with Klinefelter syndrome often will have elevated follicle-stimulating and luteinizing hormone levels and low plasma testosterone levels. Subclinical values on laboratory indices may be found in these patients; therefore, a karyotype with instructions to count sex chromosomes in 50 cells is useful for diagnosis, especially in patients with mosaicism.
Causes of Klinefelter Syndrome
The additional sex chromosomes in men with Klinefelter syndrome results from non-disjunction during meiosis and may have a paternal (50 to 60 percent) or maternal (40 to 50 percent) origin. This contrasts with Down syndrome, which is caused predominantly by maternal nondisjunction and inheritance of the extra chromosome 21 from the mother.
Although Klinefelter syndrome encompasses the specific clinical consequences observed in men with an XXY karyotype, there are variant karyotypes that can include additional X chromosomes (e.g., XXXY, XXXXY) or additional X and Y chromosomes (e.g., XXYY). Men with these karyotypes have similar but often more severe phenotypes compared with those with the XXY karyotype.
Physical manifestations of Klinefelter syndrome are relatively moderate compared with autosomal trisomies (e.g., Down syndrome) because when additional X chromosomes are present, one is predominantly inactivated. However, the entire X chromosome is not inactivated. As the number of X chromosomes increases, the phenotypic severity increases as well. As a result, cognitive and gonadal development is impaired, and cardiovascular and skeletal manifestations often are present.
Mosaicism occurs in 15 percent of men with an additional X chromosome and generally results in a milder phenotype. When mosaicism occurs, cells with two or more karyotypes are distributed. Most often, a normal chromosome number (46,XY) is identified in cells from a sample that also contains hyperdiploid cells (i.e., a Klinefelter or Klinefelter variant karyotype). In standard venipuncture, however, the ratio of normal to hyperdiploid white blood cells is not reflective of all tissues, and the phenotypic severity cannot be anticipated.
Management
As soon as a patient has been diagnosed with Klinefelter syndrome, a comprehensive neurodevelopmental evaluation is recommended. In infancy and early childhood, this may include a thorough multidisciplinary developmental evaluation to determine appropriate treatments such as physical therapy, infant stimulation programs, and speech therapy.
Men with Klinefelter syndrome have congenital (primary) hypogonadism, which results in the inability to produce the normal amount of testosterone. Consequently, these men also have hypergonadotrophism. The prepubertal testosterone deficit results in impaired bone mineral density and skeletal muscle development. Decreased energy and libido, which are associated with postpubertal testosterone deficit, improve with hormone therapy and often are accompanied by improved confidence and sense of well-being.10
Androgen therapy should be started when there is direct laboratory evidence of a testosterone deficit or when hypergonadotrophism, which suggests such a deficit, is present. This may occur by the time the patient begins middle school (i.e., 12 to 14 years of age). Physical differences in males with Klinefelter syndrome often are evident after the onset of puberty. However, the characteristic phenotypic findings (e.g., lack of gonadal development; sparse or absent facial hair; and thin, long-limbed body habitus) usually are subtle. Consideration of Klinefelter syndrome as a diagnosis and investigation with a standard karyotype are important because of the therapeutic benefit of testosterone supplementation. Clinical suspicion warrants diagnostic investigation to ensure appropriate surveillance for testosterone deficit.
Infertility in men with Klinefelter syndrome is caused by a precipitous drop in sperm count.11 If sperm are present, cryopreservation is useful for future family planning with intracytoplasmic sperm injection, and if not, testicular sperm extraction may be pursued.12 Although there have been multiple reports of successful fertilization by men with Klinefelter syndrome, genetic counseling is essential because of the increased risk of autosomal and sex chromosome abnormalities.13
Resources
The following education, support, referral, and research Web sites are useful for physicians and patients with Klinefelter syndrome:
American Association for Klinefelter Syndrome Information & Support (www.aaksis.org)
Klinefelter Syndrome Support Group (http://klinefeltersyndrome.org)
Klinefelter Syndrome and Associates (http://genetic.org/ks)
Genomics Glossary
Autosomes: Chromosomes that are not sex chromosomes. Autosomes make up the first 22 chromosome pairs.
Hyperdiploid: Having more than 46 chromosomes in one cell.
Mosaic or mosaicism: The presence of two or more cell lines. Mosaicism in a man with Klinefelter syndrome may consist of a cell line with a 47,XXY karyotype and another one with a normal number of chromosomes (46,XY).
Nondisjunction: Failure of a chromosome pair to separate properly (e.g., two X chromosomes into daughter cells).
Sex chromosomes: X and Y chromosomes (in contrast with autosomal chromosome pairs 1 through 22).